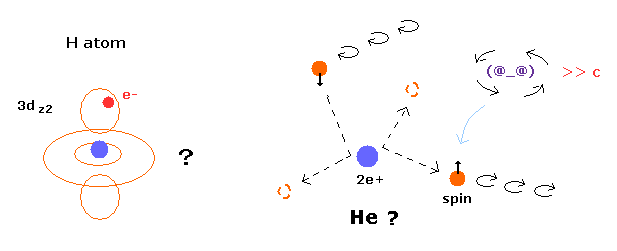
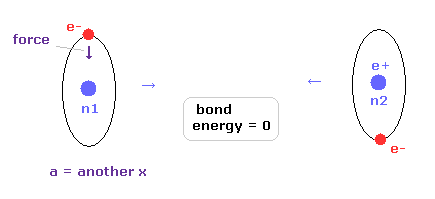
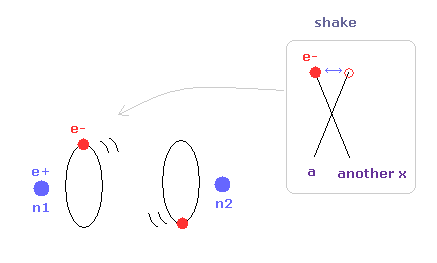
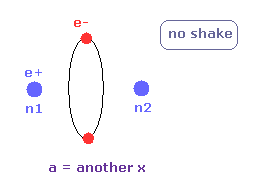
トップページ (2電子原子も含む正確な新ボーア模型)
電子スピンは現実には存在しない。
特殊相対論は間違い。
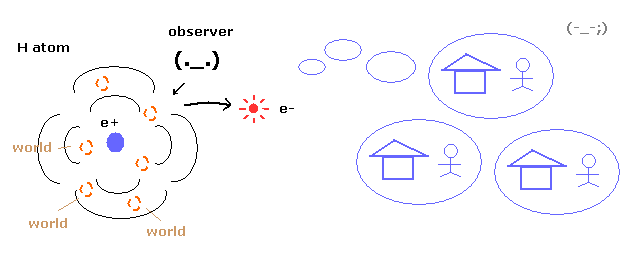
(Fig.1) 重ね合わせ = 非現実的な多世界 !
ご存じのとおり 量子力学は 電子の具体的な運動をまったく示すことができない。
それは 水素原子における 電子の確率密度のみを示すだけである。
そのため 波動関数の 収縮 (収束) を説明するためには、非現実的な多世界を必要とする。
(Fig.2) 1 × ドブロイ波長 = 水素の基底状態。
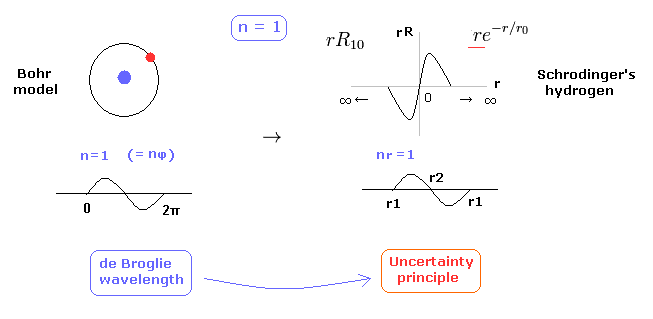
このページに示したように、シュレディンガーの水素原子も ドブロイ波長の整数倍の条件を満足する。
これが シュレディンガーの水素原子が ボーア模型と完全に同じ結果を与える理由である。
Fig.2 に示すように、水素原子の基底状態では ボーア模型は 接線方向のドブロイ波で シュレディンガーの水素 ( rR ) は 動径方向のドブロイ波で構成されている。
( 確率密度 |rR|2 のグラフを見ても このことを容易に理解できる。)
実際に、水素原子の基底状態の確率密度は ボーア半径周囲で もっとも高い。
問題は シュレディンガーの動径方向の波動関数は 必ず ゼロから無限大に広がっていなければならない。
また 1s のドブロイ波は 線状なため、それらの逆位相どうしが 重なり合って互いに打ち消しあってしまう。
(Fig.3) シュレディンガーの波動関数は 必ず無限大まで広がっている。
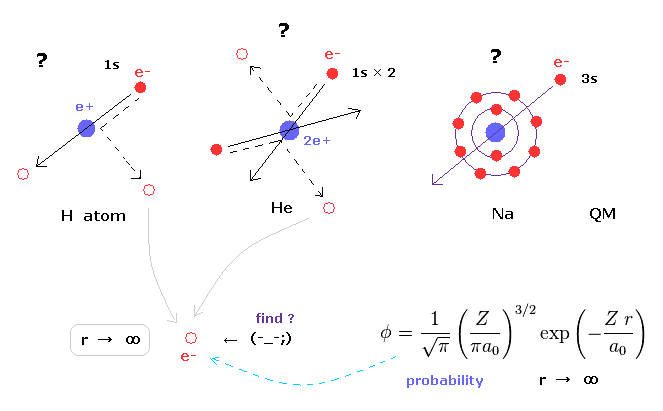
ヘリウムや水素分子なども含めた 様々な原子、分子における シュレディンガーの波動関数は 必ず 無限大まで広がっている。
これはつまり 非常に安定なヘリウムの 基底状態の電子でさえ、無限遠の近くで見つけることができるという意味である。
もちろん こんなことはあり得ないので 量子力学(化学)は 様々な原子や分子のリアルな状態を説明できる能力がないことになる。
ヘリウム原子のシュレディンガー方程式は解くことができないが、その変分関数は 必ず無限大まで広がっている。
(Fig.4) シュレディンガーの 2P の "動径方向" の波動関数 ( 角運動量 = 1 )。
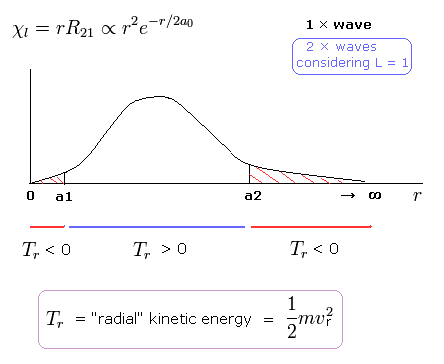
例えば、シュレディンガーの 2P の状態では、Fig.4 に示すように 動径方向の運動エネルギー (= 1/2mv2 ) が 両サイドで マイナスになる。
( 2P 状態では、 エネルギー準位は n = 2 で 角運動量は L = 1 である。 )
この奇妙な状態は いわゆるトンネル効果とは何の関係もない。
なぜなら Fig.4 の 0 から a1 の領域では、位置エネルギーは 全エネルギーよりも低いからである。
接線方向の運動エネルギーの増加をキャンセルするために、動径方向のエネルギーが マイナスにならなければならないのである。
これらの不合理な状態が シュレディンガーの水素原子が 間違いであることを示している。
(Fig.5) 量子力学のヘリウムは カオスになり不安定である。
量子化学では もし 水素原子の 1s の波動関数を使用すると、ヘリウムの近似的な基底状態のエネルギーを得ることができる。
お気づきのとおり、1s の状態は 角運動量がゼロである。
この状態では ヘリウムの2つの状態は カオス状態になり、それらの間のクーロン反発力のため 非常に不安定になる。
つまり 量子力学のヘリウムは リアルなヘリウム原子とは まったく異なるものである。
(Fig.6) 量子力学のヘリウム。
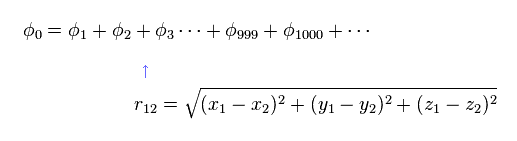
シュレデインガー方程式を用いて ヘリウム ( もしくは 水素分子 ) などの 正確な基底状態を得るためには Fig.6 に示すように 千以上の変分関数を使用しなければならない。
(Fig.7) ヘリウム もしくは H2 の確率密度 ?
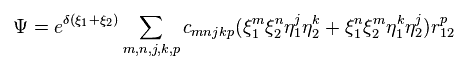
これらの変分関数は 2電子間の距離を表す変数 (= r12 ) を含まなければならない。
そのため 定常な水素原子と異なり、ヘリウム (もしくは H2 ) の電子 1 を発見する確率密度は 電子 2 の位置に応じて絶えず変化していることになる。
つまり このヘリウムの変分関数は 曖昧な確率密度波の点からも 2つの電子が実際に動いていることを示している。
(Fig.8) 換算質量 = 電子と原子核は実際に動いている。
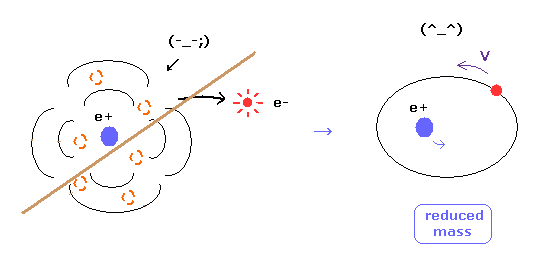
ご存じのとおり、電子の換算質量を用いると、水素原子においても より正確な エネルギー準位の値を得ることができる。
この事実は 明確に電子と原子核が互いに相互作用しながら実際に動いていることを示している。
もちろん、電子の分数電荷は 実際に見つかっていないため、ヘリウムの2つの電子のリアルな動きを直に示す必要がある。
このページに示したように、量子力学では ガイド波 (ボーム) 理論でさえ 2電子のヘリウムを説明できない。
(Fig.9) 換算質量を用いた計算結果は より正確になる。
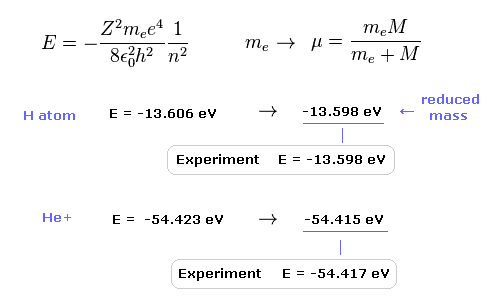
水素原子の基底状態エネルギーの実験値は -13.598 eV である。
この基底状態のエネルギーをシュレディンガー方程式 ( もしくは ボーア模型 ) を用いて計算すると、
それは -13.606 eV になる。
原子核の運動を考慮して 通常の電子質量を 換算質量に置き換えると、この計算結果は より正確になる (= 13.598 eV )。
この結果は 明らかに 原子内の電子と原子核が 実際に動いていることを示している。
He+ イオンにおいても、換算質量を用いると、より正確な値 (= -54.415 eV ) を得ることができる。通常の電子質量だと -54.423 eV となる。
He+ の実験値は -54.417 eV である。
(Fig.10) 2電子原子モデル ( He, Li+, Be2+, B3+, C4+ ... )
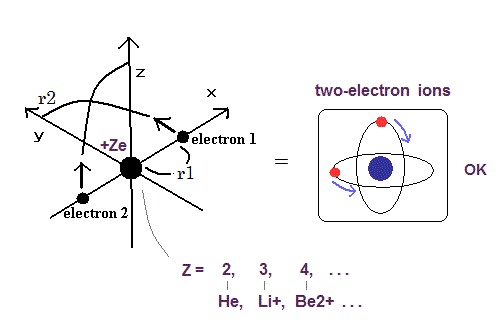
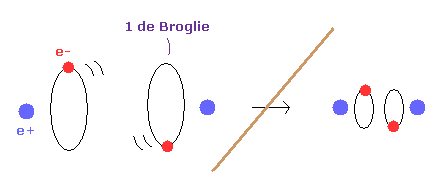
トップページに示したように、もし 2つのドブロイ波 (= 1 × 波長 ) が 互いに重なりあっているとすると、それらの逆同士の位相は干渉で打ち消し合う。
( この電子のドブロイ波の打ち消し合いは ダビッソン・ガーマーの実験で確認されている。)
そのため 2つの電子軌道は 打ち消し干渉を避けるために 互いに垂直にならなければならない。
| 原子 | r1 (MM) | WN x 4 | 同一円軌道 | 計算結果 (eV) | 実験結果 (eV) | 誤差 (eV) |
|---|---|---|---|---|---|---|
| He | 3074.0 | 1.000000 | -83.335 | -79.0037 | -79.0051 | 0.001 |
| Li+ | 1944.5 | 1.000000 | -205.78 | -198.984 | -198.093 | -0.89 |
| Be2+ | 1422.0 | 1.000000 | -382.66 | -373.470 | -371.615 | -1.85 |
| B3+ | 1121.0 | 1.000000 | -613.96 | -602.32 | -599.60 | -2.72 |
| C4+ | 925.0 | 1.000000 | -899.67 | -885.6 | -882.1 | -3.50 |
| N5+ | 788.0 | 1.000000 | -1239.8 | -1223.3 | -1219.1 | -4.20 |
| O6+ | 685.3 | 1.000000 | -1634.38 | -1615.44 | -1610.70 | -4.74 |
| F7+ | 607.3 | 1.000000 | -2083.3 | -2062.0 | -2057.0 | -5.00 |
| Ne8+ | 544.5 | 1.000000 | -2586.7 | -2563.0 | -2558.0 | -5.00 |
Table 1 に示すように、これらの2電子原子模型は 様々な原子における 実際のエネルギー状態を完全に再現することが可能である。
このヘリウムの基底状態エネルギー ( -79.0037 eV ) は 量子力学的な変分法 ( -79.015 eV ) よりも より正確な値を出すことができた。
なぜなら 量子力学のヘリウムは 原子核の動き (換算質量) を正確に扱えないからである。
具体的な計算プログラムに関しては このページを参照のこと。
(Fig.11) 新しいボーア模型のヘリウム (= A.) は 電気的に分極していない。
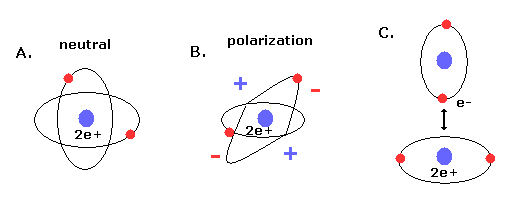
さらに このヘリウム模型は ヘリウムが最も安定な原子で かつ 他の原子 もしくは 自分自身と化合物を形成しない という事実と ちょうど一致する。
なぜなら 2つの軌道が 互いに垂直だと、2e+ 原子核の周囲の空間は 電子の均等分布のため ちょうど中性になるからである。
また ドブロイ波の安定性を考慮すると、Fig.10 のヘリウムには 3番目の電子が入っていく空間は残っていない。(= パウリの排他原理 )。
もし 3番目の電子が そこに入っていくと ドブロイ波どうしで 打ち消し合うものがでてきて 不安定になってしまうからである。
(Fig.12) "数学上の" シュレディンガー方程式 vs. "リアルな" ボーア模型。
私達は シュレディンガーの波動関数が何なのかを知ることができず、それが何かを問うことを諦めている。
さらに、このページに示したように、たとえ 相対論的な場の量子論を勉強したとしても ミステリアスなパウリの排他原理が何なのか知ることができない。
彼らは 数学上の演算子の反交換性が パウリの排他原理を意味しているという主張しかできない。
このページに示したように、私たちは この非常に制限された 曖昧な条件のもとでは より簡単で有用な方法を試して発展させることができない。
この役になっていない現在の量子化学を 分子生物学や ナノテクノロジーなどの他分野に 応用することができないため、それらの発展は 分子レベルにおいて 現在 すべてストップしている。
(Fig.13) 密度汎関数法 = 近似。
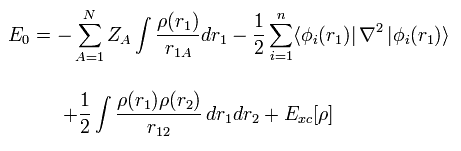
最初に、現在頻繁に使用されている 密度汎関数法 (= DFT ) は 単なる近似 であり、第一原理 ( ab-initio 方法 ) ではないということである。
DFT は 半経験的な手法の1つである。
実験結果に合わせるため LDA などの様々な近似方法を 人為的に選択しているだけにすぎない。
つまり 現在人気がある DFT そのものは 真の理論ではない。
(Fig.14) 量子力学の水素分子 ( H2 ) ?
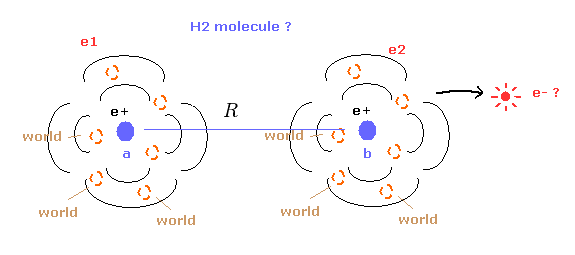
最初に 水素分子 (H2) に対する量子力学のアプローチについて知る必要がある。
水素原子と異なり、H2 分子の波動関数そのものは 電子のリアルな波動関数を意味していない。
そのため 量子力学の H2 分子から 2つの電子のリアルな状態を知ることはできない。
( もちろん この H2 の構造を他の分子に応用することもできない。)
(Eq.1) H2 分子のハミルトニアン。
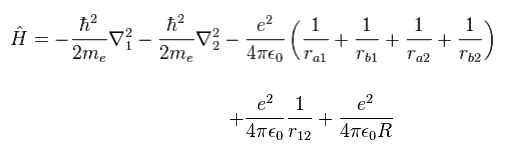
Eq.1 は 2つの電子 ( 1 と 2 ) と 2つの原子核 ( a と b ) の間のクーロン相互作用を含んでいる。
そのため この式は解くことができず、何らかの近似に頼らざるを得ない。
量子化学では、パウリの排他原理は 行列式で表現する。
なぜなら 行列式内では 各波動関数の状態は 互いに異なっていなければならないからである。
(Eq.2) H2 の波動関数 = 行列式 ?
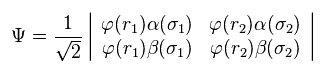
Eq.2 では φ は 軌道の波動関数、α β は スピン状態を表している。
もちろん 多電子原子や分子では、この行列式は 次のようにかなり複雑なものになる。
(Eq.3)
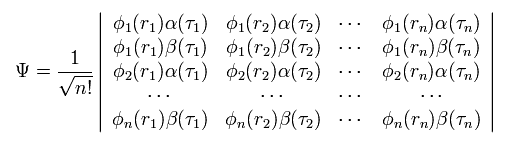
これら複雑な数学的表現のために 私達は 現在の量子化学を 様々な分野に応用することができない。
しかし 量子力学は 奇妙な スピンに関して ただ "Shut up and calculate !" を繰り返すだけなので パウリの排他原理を表す他の方法がないのである。
(Eq.4) 各波動関数。
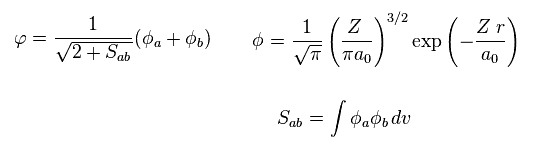
ここで a0 は ボーア半径である。
各電子の波動関数は 原子核 a と b に属する状態を含んでいる。
そして 水素様原子 の 1s 波動関数を使用する。
( Z は 原子番号である。水素原子では Z = 1 になる。)
Eq.4 を Eq.2 に代入すると、
(Eq.5)
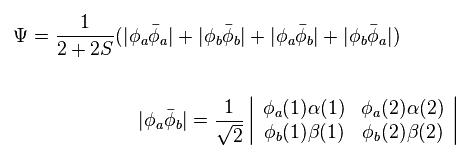
Eq.1 と Eq.5 を使用して、次の波動関数を解く。
(Eq.6)
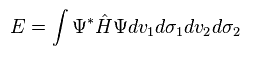
もっとも単純な H2 分子でさえ、この計算は複雑である。
計算結果は、
(Eq.7)
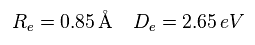
Re は 2つの原子核の距離である。De は H2 の 解離 (結合) エネルギーである。( H2 → 2H ).
Eq.7 の結果は 次の実験結果と異なる。
(Eq.8) H2 の実験値。
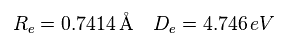
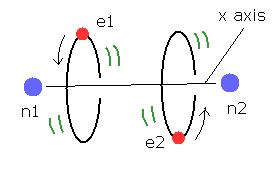
原子価結合 (VB) 法と分子軌道法 (MO) の両方において、正確な値を得ることができない。
もっと正確な値を得るためには、もとの波動関数に架空のイオン状態 ( H2 = H- - H+ ) を 付け加える必要がある。
(Eq.9)
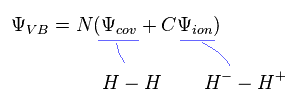
このアプローチでさえ、計算結果は 約 3.5 eV で、実験値と異なる。
また もちろん、実際の水素分子では このイオン状態は起きていない。
つまり これらの人為的な方法は 計算上の道具にすぎず、本当の状態を表していない。
変分法の真実も参照のこと。
(Eq.10) 本当の水素分子? = 100 項以上。
ここで δ と c は 変分関数である。
正確な値を得るためには、r12 などの変数を含んだ 非常に込み入った波動関数を解かなければならない。
" r12 " は 2つの電子間の距離を意味する。
そのため 電子1を ある場所で見つける確率密度は 電子 2 の位置に応じて変化していることになる。
つまり H2 分子や ヘリウム原子では 確率密度そのものを決定できないことになる。
また Eq.10 において 項の数や種類を 人為的に選ぶことが可能であるため、彼らは 無限種類の選択枝の中から 実験値を与える波動関数を選んでいるだけである。
もちろん、私達は Eq.10 の非常に込み入った数学上の (物理でなく) 項を 分子生物学や何かの他分野に応用することができない。
また 上で述べたように、この波動関数は必ず 無限遠まで広がっており、リアリティーがない。
(Fig.15) 推定される H2 分子。
核間距離 ( 0.7414 × 10-10 m ) と 2つの電子と原子核間のクーロン相互作用を考慮すると、H2 分子の実際の電子状態は Fig.15 のようになると考えることのは自然である。
もちろん、クーロン相互作用は 量子化学においても もっとも重要な概念ある。
また 分子の形を決定する他の重要な概念が ドブロイ波長である。
(Fig.16) もし ドブロイ波長が存在しないと?
もし ドブロイ波長の概念がないと、水素原子の電子軌道は ゼロにまで小さくなれる。
結果的に H2 分子の核間距離は 好きなだけ短くなることが可能である。
なぜなら 2つの核間の反発力が 任意に小さな電子軌道によってキャンセルされるからである。
つまり ドブロイ波長の概念によって H2 の核間距離 ( 0.7414 × 10-10 m ) が保たれていることになる。
(Eq.11) シュレディンガーの波動関数もドブロイ波長を含んでいる。
ここで a0 は ボーア半径である。
実際には 量子化学においても、このドブロイ波長の概念は自動的に使用されている。
このページに示したように、シュレディンガーの 1s の波動関数は 動径方向に 1 × ドブロイ波長である。
( 彼らは ドブロイ波長の名前を 奇妙な不確定性原理に変えただけである。)
原子番号 Z を変分関数として変化させると、様々な相互作用においても この 1 × ドブロイ波長の条件を保つことができる。
( 例えば、H → He+ イオン の変化では、両方の基底状態とも 1 × ドブロイ波長である。)
(Fig.17) ドブロイ波長と 半径。
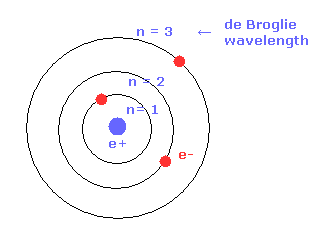
ボーア軌道に示したように、ドブロイ波長の数が 多くなるにつれて 電子軌道の半径は 長くなる。
例えば、2 × ドブロイ波長の軌道半径は 1 × ドブロイ波長のものの 4倍である。
( n = 1 -- 1 × ボーア半径、 n = 2 -- 4 × ボーア半径。 )
このボーア半径の概念は 量子力学においても有効である。
(Fig.18) ドブロイ波長と電荷。
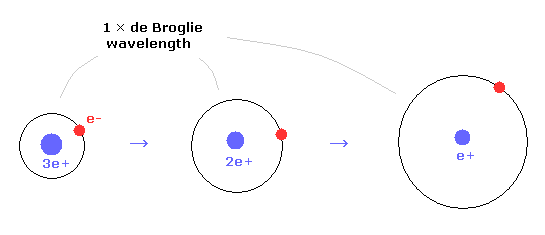
1 × ドブロイ波長では、 中心の正電荷が小さくなるにつれて 軌道半径が 長くなっていく。
例えば、ヘリウムイオンの半径は 水素原子の半分である。
これらの概念とクーロン相互作用を組み合わせると、H2 分子の 原子間の結合を説明することができる。
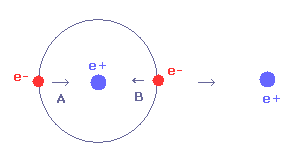
(Fig.19) H2+ 分子イオンの2つの原子核。
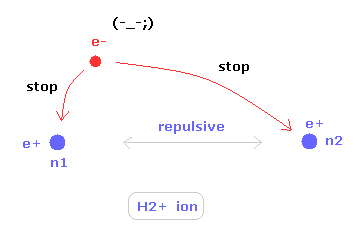
ある特殊な条件下で、不安定な水素分子イオン (H2+) が存在できる。
水素分子イオンは 2つの正の原子核と 1つの負の電子を持つ。
2つの原子核の位置を安定に保つために、この唯一の電子は 水素分子に比べて 非常に活発に動き回る必要がある。
そのため、H2+ 分子イオンは ヘリウム原子とは まったく異なるものである。なぜなら 電子は その自身の運動のみによって 2つの原子核を安定に保つ必要があるからである。
この余分な運動 (= ドブロイ波 ) は 分子において非常に重要である。
(Fig.20) H2 分子の 2つの原子核。
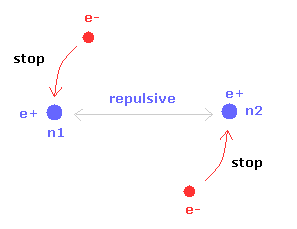
H2+ 分子と異なり、H2 分子は 2つの電子を持つ。
そのため 各電子は 2つの原子核の どちらか一方のみをメインに保つように運動している。
2つの原子核のうち片方が安定になると、もう1つの原子核は 対称的な構造のために 自動的に安定になる。
結果的に H2 分子の電子は H2+ 分子イオンのように 激しく揺れ動く必要がない。
しかしもちろん、原子核と電子そのものの安定位置は 異なる。
そのため この余分な動きを H2 分子においても 考慮する必要がある。
(Fig.21) H2 分子。
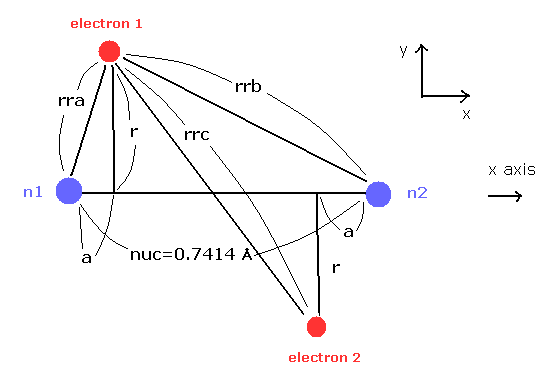
ここで Fig.21 の模型と 次のプログラムを用いて 1軌道に含まれる ドブロイ波の数を計算することにする。
H2 分子のサンプル JAVA プログラム。
H2 分子の C 言語プログラム。
ここでは 1 MM = 1 × 10-14 meter の新しい単位を使用する。
このプログラムでは、最初に H2 の核間距離 (= nuc, MM ) を入力する。
その後、電子1の y座標 "r" ( MM )、x 座標 "a" ( MM ) を 入力する。
(Fig.22) ( a, r ) における様々な力。
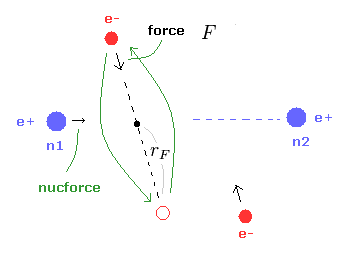
これら入力値から、このプログラムは H2 の結合エネルギー (eV)、原子核 1 に作用する トータルの力 (= nucforce )、電子1に作用する力の x, y 成分 (= elefx, elefy ) を出力する。
これらの力は 水素原子の基底状態の電子と原子核間の力との比で表されている。
(Fig.23) e1 に作用する 力 F。
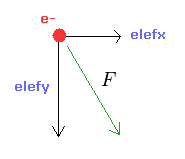
入力値 ( a, r, nuc ) より、全位置エネルギー V を計算する。
ビリアル定理を用いて、全エネルギー E = 1/2 V と 運動エネルギー T = -1/2 V を得る。
この運動エネルギーより、電子速度 (= v ) と ドブロイ波長 (= h/mv ) を計算する。
(Eq.12) F = 遠心力。
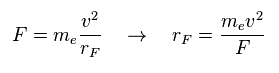
ここでは 力 F を何らかの遠心力と仮定する。
Eq.12 の関係式から 回転半径 (= rF ) を得ることができる。
(Eq.13) 1軌道のドブロイ波
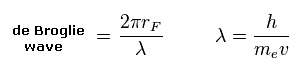
ドブロイの関係式 ( λ = h/mv ) を用いて、1軌道 (= 2πrF ) に含まれるドブロイ波の数を計算する。
ビリアル定理を用いて、位置 ( a, r ) における H2 分子の 結合エネルギーを計算する。
(Fig.24) 電子の もう1つの x 座標。
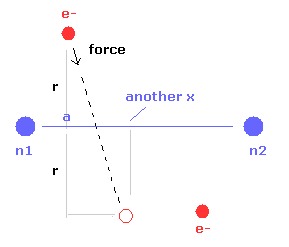
また このプログラムは Fig.24 に示すように 電子1が 力 F の方向へ動いたときの もう1つの x 座標 ( MM ) を出力する。
この x 座標は 電子の x 方向への揺れの指標になる。
最初に 結合エネルギー (= 4.746 eV ) と 核間距離 (= 0.7414 × 10-10m = 7414 MM ) が 実験値のケースについて考える。
(Eq.14) H2 の実験値。
電子1が ( a = 624 MM, and r = 4660 MM ) にあるとき、原子核に作用する 力の合計はゼロになる。
( nucforce = 0。 この地点で 2つの原子核は 安定になる。 )
(Fig.25) 実験値。
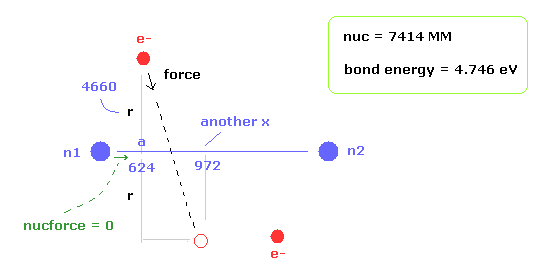
この地点において、1軌道に含まれる ドブロイ波の数 (= Eq.13 ) は0.977 になる。
この値は だいたい 1 になるが 若干異なる。
なぜなら電子軌道は x 方向に少し揺れるからである。
もう1つの x 座標 ( a, r ) = ( 972, 4660 ) のとき、このドブロイ波は 1.009 になる。
結果的に 平均のドブロイ波は ( 0.977 + 1.009 ) / 2 = 0.993 となる。
この値は "1" に近くなる。
| nuc (MM) | r (MM) | a (MM) | Energy (eV) | another x | nucforce | waves | aver waves |
|---|---|---|---|---|---|---|---|
| 6000 | 4770 | 1470 | 4.341 | 1689 | 0.00 | 0.977 | 0.985 |
| 6500 | 4720 | 1030 | 4.546 | 1329 | 0.00 | 0.977 | 0.991 |
| 7000 | 4680 | 770 | 4.698 | 1105 | 0.00 | 0.977 | 0.993 |
| 7414 | 4660 | 624 | 4.746 | 972 | 0.00 | 0.977 | 0.993 |
| 8000 | 4650 | 480 | 4.705 | 830 | 0.00 | 0.977 | 0.992 |
| 8500 | 4640 | 390 | 4.689 | 728 | 0.00 | 0.977 | 0.990 |
| 9000 | 4642 | 325 | 4.602 | 645 | 0.00 | 0.977 | 0.989 |
Table 2 に示すように、共通な条件 nucforce = 0 と ドブロイ波 = 0.977 を選ぶとき、核間距離 nuc = 7414 MM (= 実験値 ) のときの 結合エネルギーが 最も大きくなる。
すでに述べたように、安定な原子核と ドブロイ波長を考慮すると、水素原子の 最も安定な状態が決定する。
平均のドブロイ波 (= aver waves ) も nuc = 7414 MM のときに最も大きくなる。
( 1 MM = 1 × 10-14 meter )
もし aver waves = 0.993 を共通条件として選ぶと、これらの結合エネルギーの差は ますます広がる。
結果的に nuc = 7414 MM のときが最も安定となる。
| nuc (MM) | r (MM) | a (MM) | Energy (eV) | elefx | elefy | nucforce | waves |
|---|---|---|---|---|---|---|---|
| 7414 | 4665 | 890 | 4.746 | 0.00 | 1.271 | 0.08 | 1.001 |
電子に作用する力の x 成分がゼロのとき ( elefx = 0.00 )、ドブロイ波は ちょうど 1.001 になる。
nuc = 7414、 r = 4665、 a = 890 ( これは Table 2 の 624 と 972 の間である ) を代入してみるといい。
(Fig.26) 2つの H 原子が 完全に分離されている。
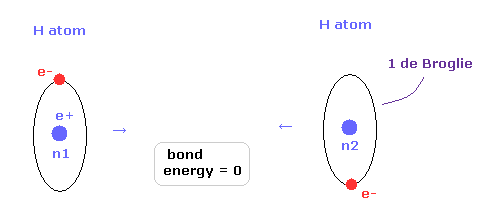
次に 核間距離が nuc = 7414 MM (= 実験値 ) のとき どうして H2 分子が最も安定になるのかという理由について考える。
Fig.26 では、2つの水素原子は 完全に分離しているため、結合エネルギーは ちょうどゼロである。
(Fig.27) 最も安定な H2 の形。
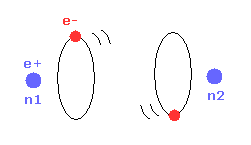
2つの原子核が互いに近づくにつれて、各電子は 2つの正の原子核と相互作用することができる。
そのため 結合エネルギーが大きくなり 安定な H2 分子が形成される。
(Fig.28) 2電子間の反発力。
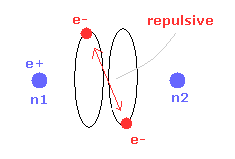
しかし 2つの電子が 実験値 7414 MM よりもさらに近づくと、2つの電子間の反発力が さらに強くなる。
結果的に Fig.18 の "実質的"な中心電荷が小さくなる。
そのため 共通のドブロイ波の条件のもとで、半径 "r" は 大きくなり、結合エネルギーは 実験値よりも小さくなる ( Table 2 参照のこと )。
(Fig.29) 核間距離が短くなると・・
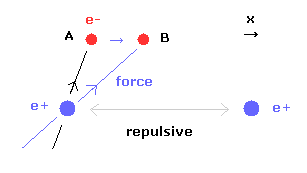
核間距離が短くなると、原子核間の反発力が強くなる。
この反発力をキャンセルするために、2つの電子は 中心方向に移動する必要がある。
Fig.29 に示すように、電子1が x 方向に動くと ( A → B )、電子1による x 方向の引力が強くなる。
しかし この場合 Fig.28 に示すように 2つの電子間の反発力が強くなる。
(Fig.30) "another x".
すでに述べたように、 "another x" は 電子1が 力の方向に動いたときの もう1つの x 座標である。
例えば ( a, r, nuc ) = ( 624, 4660, 7414 ) のとき、 another x は 972 (MM) になる。
次に 同じ核間距離で 座標 ( anotherx, r ) のときの ドブロイ波と結合エネルギーを計算する。
つまり Fig.30 のケースでは、 ( a, r, nuc ) = ( 972, 4660, 7414 ) の条件で再び計算する。
そして 元の "a" と "another x" のときの、平均の結合エネルギーとドブロイ波をだす。
| nuc (MM) | r (MM) | a (MM) | AveEnergy (eV) | another x | nucforce | waves | aver waves |
|---|---|---|---|---|---|---|---|
| 6500 | 4744.3 | 1059 | 4.408 | 1351 | 0.00 | 0.9797 | 0.9933 |
| 7000 | 4684 | 775 | 4.689 | 1107 | 0.00 | 0.9773 | 0.9933 |
| 7200 | 4671 | 697 | 4.732 | 1037 | 0.00 | 0.9771 | 0.9933 |
| 7414 | 4662.3 | 629 | 4.746 | 972 | 0.00 | 0.9772 | 0.9933 |
| 7600 | 4658 | 577 | 4.738 | 922 | 0.00 | 0.9774 | 0.9933 |
| 8000 | 4657 | 488 | 4.671 | 831 | 0.00 | 0.9784 | 0.9933 |
| 8500 | 4665.3 | 402 | 4.532 | 739 | 0.00 | 0.9797 | 0.9933 |
Table 4 では、原子核に作用する力がゼロ ( nucforce = 0.00 )、 平均のドブロイ波が 0.9933 を共通の条件に選んだ。
これらの条件では、Table 3 のように、 nuc = 7414 MM (= 実験値 ) のとき 結合エネルギーが 最も大きくなる (= 4.746 eV ).
結果的に 核間距離が 0.7414 × 10-10 meter のとき 最も 安定になる。
JAVA サンプルプログラム H2 分子 ( 平均値 )。
C 言語 H2 分子 ( 平均値 )。
上記のプログラムを利用すれば、1軌道のドブロイ波の平均値を自動的に計算してだす。
この場合も同様に "nuc"、 "r"、 "a" (MM) ( Table 4 参照 ) の値を最初に入力する。
(Fig.31) 結合エネルギーがゼロ → "a" = another x。
"another x" の座標も考慮する理由が Fig.31 に示してある。
核間距離が非常に長いとき、結合エネルギーはゼロになる。
この場合は、各水素原子は独立になり、その軌道は もう1つの原子核によって 揺れ動かない。
つまり Fig.31 では 元の x 座標 "a" は ちょうど "another x" に等しく、 Table 2 のように 平均でないドブロイ波 waves = 0.977 を共通条件として選ぶと、正しいエネルギー計算結果をだせない。
(Fig.32) "a" が another x と異なる。 = 軌道が揺れている。
核間距離が Fig.31 よりも短くなると、もう1つの原子核の影響が強くなり 電子軌道が揺れ動くことになる。
この場合でとくに、平均のドブロイ波が非常に重要になる。
(Fig.33) また "a" = another x になる。
さらに核間距離が短くなっていくと、電子軌道はまた揺れなくなる。
この場合も "a" が "another x" にちょうど等しくなる。
( もちろん 2つの電子は 1 × ドブロイ波長の同一軌道に入れないが。)
つまり 最も安定な形のとき、電子軌道は 横に揺れていることになる。
(Fig.34) 1軌道内の 2つの力は異なっている。
例えば Fig.34 では、同じ軌道内ではあるが、力 B は 力 A よりも弱い。
もちろん B においては 位置エネルギーは低く、よって ビリアル定理によれば ドブロイ波長は短くなる。
つまり B のところで ( Eq.12, Eq.13 を用いて ) 計算すると、1軌道内のドブロイ波の数は 平均値よりも大きくなってしまう。
そのため A と B の平均値をとる必要がある。
上のセクションで、ボーア軌道を用いて 水素分子を表すことに成功した。
このページでは このプログラム もしくは そのテキスト を用いて 水素分子に関して復習する。
( このページも参照のこと。 )
プログラム実行後、A、B atom 両方において "H" を選択する。
H2 の核間距離は 0.7414 Å であるため、7414 を入力して "internuc" ボタンをクリックする。
(Fig.35) H2 分子。
このプログラムでは、A と B 原子の両方において "ele 0" の x と z 座標を対称的に変化させる。
ele 1 は ele 0 に作用する力で 動いた後の 電子の座標である。
A と B 原子核の両方で安定 ( FX = 0 ) と 平均のドブロイ波が "1.0000" ( ave = 1.0000 ) であることを目指す。
| 核間距離 (MM) | A-X (MM) | A-Z (MM) | B-X (MM) | B-Z (MM) | 核への力 | ドブロイ波平均 | total V (eV) | 結合エネルギー |
|---|---|---|---|---|---|---|---|---|
| 6000 | 1870 | 4972 | -1870 | -4972 | 0 | 1.0000 | -60.664 | 3.120 |
| 6500 | 1145 | 4827 | -1145 | -4827 | 0 | 1.0000 | -62.204 | 3.890 |
| 7000 | 830 | 4755 | -830 | -4755 | 0 | 1.0000 | -62.893 | 4.234 |
| 7200 | 745 | 4739 | -745 | -4739 | 0 | 1.0000 | -63.014 | 4.295 |
| 7414 | 669 | 4729 | -669 | -4729 | 0 | 1.0000 | -63.056 | 4.316 |
| 7600 | 610 | 4724 | -610 | -4724 | 0 | 1.0000 | -63.047 | 4.311 |
| 7800 | 560 | 4721 | -560 | -4721 | 0 | 1.0000 | -63.010 | 4.293 |
| 8000 | 518 | 4720 | -518 | -4720 | 0 | 1.0000 | -62.952 | 4.264 |
| 9000 | 360 | 4741 | -360 | -4741 | 0 | 1.0000 | -62.365 | 3.971 |
H2 の平均の結合エネルギーは 最後の行 (= "bin" ) に示してある。
Table 5 の total V は 平均のポテンシャルエネルギーである。
Table 5 に示すように、H2 分子の結合長が 実験値 0.7414 Å (= 7414 MM ) のとき、 total V は もっとも低くなり、結合エネルギーは 最も高く (= 4.316 eV ) なる。
この値は 実験値 4.746 eV よりも少し小さい。
しかし この結果は H2 結合長が 0.7414 Å であることが 古典力学的観点から明らかに説明できたことを示している。
| 核間距離 (MM) | A-X (MM) | A-Z (MM) | B-X (MM) | B-Z (MM) | 核への力 | ドブロイ波平均 | total V (eV) | 結合エネルギー |
|---|---|---|---|---|---|---|---|---|
| 6000 | 1665 | 4878 | -1665 | -4878 | 0 | 0.9933 | -61.787 | 3.681 |
| 6500 | 1059 | 4745.5 | -1059 | -4745.5 | 0 | 0.9933 | -63.224 | 4.400 |
| 7000 | 775 | 4684 | -775 | -4684 | 0 | 0.9933 | -63.802 | 4.689 |
| 7200 | 697 | 4671.5 | -697 | -4671.5 | 0 | 0.9933 | -63.8815 | 4.728 |
| 7414 | 629 | 4662.3 | -629 | -4662.3 | 0 | 0.9933 | -63.916 | 4.746 |
| 7600 | 577 | 4658 | -577 | -4658 | 0 | 0.9933 | -63.899 | 4.738 |
| 7800 | 530 | 4657 | -530 | -4657 | 0 | 0.9933 | -63.838 | 4.707 |
| 8000 | 488 | 4657 | -488 | -4657 | 0 | 0.9933 | -63.765 | 4.670 |
| 9000 | 340 | 4681.5 | -340 | -4681.5 | 0 | 0.9933 | -63.125 | 4.350 |
次に このページに示したように1軌道のドブロイ波が 0.9933 のときを調べる。
Table 6 に示したように、H2 分子の結合長が 実験値 0.7414 Å (= 7414 MM ) のとき、 total V は 最も低く、結合エネルギーは 最も大きく (= 4.746 eV ) なる。
結合エネルギーの実験値は 4.746 eV である。
この結果は と 真の H2 分子の結果と一致する。
つまり この簡略化した方法では ドブロイ波 = 0.9933 が 正しい標準値である。
もちろん、 上記の両方の基準において H2 の結合長 = 0.7414 Å が 最も安定になることには 変わりがない。
結果的に 分子の形は クーロン相互作用と ドブロイ波長という 2つの重要な性質によって決まることになる。
これが 分子結合の本質である。
(Eq.10) 量子力学の H2。
ご覧のとおり、新しいボーア模型の水素は 量子力学の H2 の数学上の ( 物理でなく ) 項の羅列より より明白で簡単な H2 構造を与えることができる。
そのため この新しい構造を 他の分子結合に より容易に応用することができる。
最も重要な点は ボーア模型には リアリティーがあるが、量子力学的な模型には それがないということである。
水素分子イオンに関しては このページを参照のこと。

2013/2/12 updated This site is link free.