Home page
Quantum computers useless in drugs.
(Fig.1) For unsolvable Schrödinger equations, quantum mechanics has to choose fake trial wavefunctions out of infinite choices, which is also useless, time-consuming, cannot predict anything.
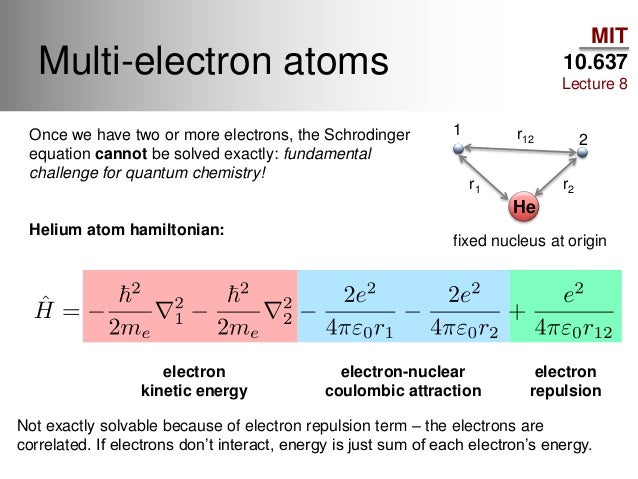
Quantum mechanics relies on the impractical Schrödinger equation consisting of electrons' kinetic and Coulomb potential energy equal to total (= Hamiltonian H ) energy E.
This Schrödinger equation, which is the only tool of quantum mechanics, is unsolvable and unable to predict energy of any multi-electron atoms or molecules.
For unsolvable Schrödinger equations, quantum mechanics can only choose fake trial wavefunctions (= fake solutions ) or basis set functions out of infinite free choices ( this-4~5th-paragraphs ) and infinite free parameters ( this-p.9-last ) to find ones giving the lowest (fake) total energy E, which quantum mechanical approximate variational methods take infinite time, so impractical.
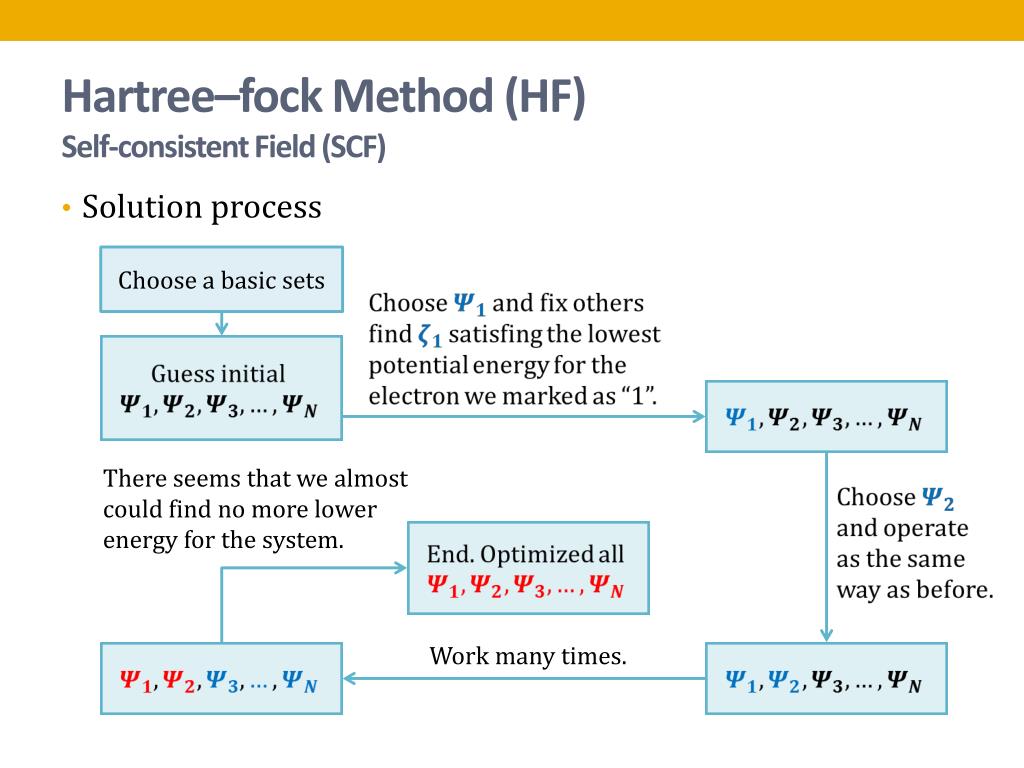
In these quantum mechanical approximate variational (= Hartree-Fock ) methods, just finding free (coefficient) parameters giving the lowest energy within chosen fake trial wavefunctions is called "(fake) solving", which is Not really solving the unsolvable Schrödinger equations
These quantum mechanical approximate variational methods for unsolvable Schrodinger equations cannot give nor predict true atomic energies ( this-p.2~p.3, this-(9.80) ). ← Because it is impossible to find the lowest energy out of infinite forms of trial wavefunctions and infinite free parameters.
This or this-p.4-1st-paragraph says -- No quantum mechanical prediction
"Notice that this (quantum approximate variational) method cannot give us an idea of difference between the upper
bound we calculate using the trial wave function and the actual ground state
energy... So we do Not have any way of
knowing how good or how bad our (chosen) trial wave function is.
The variational method also does Not tell us what kind of a trial function we must
choose"
For unsolvable Schrödinger equations that cannot predict anything, quantum mechanical approximate variational methods such as Hartree-Fock and configuration interaction (= CI ) have to choose fake trial ( antisymmetric ) wavefunctions expressed as unphysical Slater determinants out of infinite free choices.
↑ All these quantum mechanical approximate methods such as Hartree-Fock and CI are impractical, too time-consuming ( this-p.2-introduction, this-p.14-last ), cannot predict anything.
This-1.4 says -- Useless Schrodinger equation
"This wave function must satisfy the Schrödinger equation, which is the fundamental equation of quantum mechanics...
However, this approach faces a severe limitation: it becomes impossibly complicated for systems with more than a few electrons. Even with modern supercomputers, exactly solving the Schrödinger equation for a molecule as simple as ethanol is beyond our capabilities."
For unsolvable Schrödinger equations, quantum mechanics has to rely on its approximate variational methods just choosing fake wavefunctions or fake solutions out of infinite free choices and free parameters ( this-4~5th-paragraphs ).
↑ This quantum mechanical variational method is supposed to give only energy higher than the true atomic ground-state lowest energy ( this-p.3(or p.1)-lower ), which is false.
Actually physicists can get energy close to experimental values even when they just roughly use only a few free (exponent) parameters fixed to to some values even without rigorously adjusting infinite free parameters in infinite terms ( this-p.11.p.25, this-p.2-lower ). ← Quantum mechanical variational methods are wrong.
↑ To get energy close to experimental helium ground-state lowest energy (= Helium ground-state energy E = - 2.9037243770.. Hartree energy without reduced mass ), the non-linear exponent (= scaling ) free parameters ( this-p.2-(1) ) of artificially chosen fake trial wavefunctions were roughly fixed to k = 2 ( this-p.2-(0.3) ) or α = 2.20 ( this-p.4-(19), p.9-Table V. p.10-right-E. ) without rigorously adjusting those parameters to find really the lowest energy.
↑ So if they more rigorously adjust more free (exponent) parameters, they can get wrong energies lower than the true atomic ground-state lowest energy, which fact disproves quantum mechanics and its variational methods.
For the impractical unsolvable Schrödinger equation that are too time-consuming and unable to predict anything, quantum mechanics has to roughly treat the whole molecules as one fake electron DFT (= density functional theory ) model ( this-p.2-p.3, this-3rd-paragraph ).
↑ This current mainstream quantum mechanical DFT approximation has to choose fake exchange-correlation (= XC ) potential energy functionals, which are also impractical, too time-consuming, unable to predict anything ( this-3rd-paragraph, this-p.7, this-p.3, this-p.1-right-1st-paragraph ).
Overhyped unrealistic quantum computers are useless, too error-prone to give right answers, cannot factor even simple 21, much less calculate molecular energy contrary to a lot of fake news.
For today's useless error-prone quantum computers, companies such as IBM, IonQ, and universities try to make deceptive hybrid quantum-classical computer methods (= just classical computers ) such as VQE, quantum-selected configuration interaction (= QSCI ) equal to sample-based quantum dianonalization (= SQD ).
↑ In all these deceptive hybrid computer methods, actual calculations of molecular energy (= Hamiltonian ) based on empirically-chosen fake trial wavefunctions (= called ansatz ) for unsolvable Schrödinger equations were conducted by classical computers with No quantum computation nor quantum mechanical prediction.
In the recent deceptive hybrid quantum-classical computer methods called quantum-selected configuration interaction (= QSCI) equal to sample-based quantum dianonalization (= SQD ), today's useless quantum computers could only get random meaningless numbers (= sampling ) from qubits initialized by parameters empirically guessed or calculated by classical computers called " ansatz"
↑ So No quantum computation nor quantum mechanical prediction in the current hybrid quantum computation of molecular energy.
↑ These hybrid quantum-classical computer methods called QSCI = SQD are based on impractical time-consuming configuration interaction (= CI or full-CI = FCI, this-p.2-introduction ) just choosing fake trial wavefunctions (= Ψ ) consisting of multiple coefficient free parameters (= c ) and unphysical Slater determinants (= configurations ) for unsolvable Schrodinger equations, which cannot predict any energies.
Useless error-prone quantum computers are used only to randomly sample parameters for these Slater determinants (= configurations, this-p.2-Fig.1 ) from qubits initialized or empirically guessed by classical computers called ansatz. ← No quantum computation nor quantum mechanical prediction of energy
In the deceptive hybrid computing methods, classical computers have to calculate and find coefficient free parameters (= c ) giving the lowest (fake) total (= Hamiltonian ) energy of some molecules in the process called diagonalization ( this-(61), this-(7.10), this-lower-equation.56 ) within artificially-chosen trial wavefunctions by quantum mechanical variational methods, which cannot predict anything.
This-p.4 says -- No quantum computation
"The quantum selected configuration interaction (QSCI) method (= SQD ).. The central idea of the method is that some approximate ground state is prepared on a quantum
computer (= called ansatz empirically guessed first ), which is then used to draw samples corresponding to determinants in the computational basis. The CI Hamiltonian is then classically diagonalized"
"First, since the quantum device is only used for selection, while energies are obtained classically (= molecular energy must be calculated by classical computers )"
Today's useless error-prone quantum computers can do nothing except for giving random meaningless numbers.
In this deceptive hybrid computer method called QSCI equal to SQD, the more random parameters designating Slater determinants are chosen by the error-prone quantum computers, the more terms or more coefficient free parameters (= c ) adjustable by classical computers in fake trial wavefunctions are available to artificially lower the energy to desired values in the impractical quantum variational method CI that cannot predict anything.
↑ But just randomly choosing or sampling parameters of Slater determinants of the artificially-chosen fake trial wavefunctions is inefficient, taking much time to get the desired (fake) wavefunctions or energy.
So they need to artificially initialize quantum computers' qubits by some empirically-guessed parameters called ansatz from which the useless quantum computers randomly pick parameters used for (artificially-chosen fake) trial wavefunctions.
↑ So No quantum mechanical prediction nor quantum computation
This-p.2-left-C. says -- No quantum mechanical prediction
"In quantum computing, an
Ansatz refers to a (fake)
trial wavefunction (= artificially- chosen ) or trial state used as a starting point for approximations or optimizations. It is a parameterized quantum
state that serves as an educated
guess (= empirically-chosen, so
No quantum mechanical prediction ) for solving a particular
problem, such as finding the ground state of a quantum system."
" Selecting an appropriate ansatz is crucial in quantum algorithm design, as it significantly impacts the accuracy of the outcomes."
In these deceptive hybrid methods for seemingly calculating molecular energies, for unsolvable Schrodinger equation, they have to artificially choose fake trial wavefunctions called ansatz based on impractical time-consuming quantum approximation called coupled cluster (= CCSD, this-p.39 ) whose energy was calculated by classical computers ( this-p.3-Fig.2, p.4-right-2. this-p.4-Figure.1 ).
↑ No quantum computation nor quantum mechanical prediction.
This-p.2-left-last-paragraph says -- Classical computer initial guess
"Another strategy to prepare the
quantum state for QSCI is to perform the coupled cluster
singles and doubles ( CCSD ) calculation (= impractical CCSD choosing unreal non-variational wavefunctions for unsolvable Schrodinger equation that cannot predict anything ) on a classical computer
and embed the CCSD wave function using the local unitary
cluster Jastrow (LUCJ) ansatz ( this-p.9-Fig.9 )"
This-p.4-right-last~p.5 says -- Empirical, No quantum prediction
"It has been empirically observed that the parameters derived from CCSD yield good
choices to run SQD experiments in noisy quantum
processors"
The recent IBM research in 2025 also used this deceptive hybrid method called SQD (= QSCI ) to seemingly get energies of water and methane molecular dimers with No quantum computation nor quantum mechanical prediction.
↑ This IBM hybrid research paper (= this-last-link ) ↓
p.1-abstract says -- Fake hybrid quantum computer
"We use a sample-based quantum
diagonalization (SQD) approach (= just classical computer method, No quantum computation nor quantum mechanical prediction ) to simulate the potential energy surfaces (PES) of the water and
methane dimers,.. We benchmark
our quantum simulations (27- and 36-qubit circuits).. 54 qubits (= one qubit can take only 0 or 1 value, so still Not a quantum computer )"
p.4-right-2nd-paragraph says -- Classical computer guessed parameters
"Parametrization of LUCJ circuits (= empirically-guessed initial qubits' state called ansatz ) based on the t2-amplitudes,
obtained from CCSD calculations on classical computers (= quantum computer is useless ), yields electron
configuration distributions corresponding to total energies"
p.6-right-2nd-paragraph says -- No quantum computer advantage
"Finally, we would like to note that
the demonstration of quantum advantage on real quantum hardware in
electronic structure simulations, to our knowledge, has Not been achieved by
any research group yet, neither with SQD, nor any other quantum accelerated techniques and, as such, remains one of the most challenging
tasks in quantum computing"
↑ This research' reviewers debunked IBM fake quantum advantage, this ↓
p.2-last says -- Classically chosen parameters
"1. The workflow, as far as I can understand, consists of: (i) a classical calculation (CCSD on a active space = ) that (ii) informs
the choice of the parameters in the LUCJ wavefunction (= ansatz ) prepared on the quantum hardware"
p.3-1st-paragraph says -- No quantum computer advantage
"The quantum part of the algorithm (iii) is therefore used to extract (= randomly sample ) electronic configurations with a probability
related to how much they were represented in the initial wavefunction (= guessed ansatz ), that in turn was informed by the initial classical
calculation. I think it should be made clear what is the practical advantage in extracting such configurations from the
quantum hardware rather than directly from the initial classical wavefunction"
p.30-5th-paragraph says -- IBM admits No advantage
"We agree that currently we do Not have sufficient data to demonstrate the possibility of quantum
advantage in simulations of non-covalent interactions."
p.32-1st-paragraph says -- No hybrid SQD advantage
"We have now gone through the rebuttal and the revised manuscript. We still do Not see answers to
crucial questions we asked at the first review round: Why encoding the LUCJ wavefunction on a quantum
computer and extracting from there should be more efficient than extracting classically"
"In particular it is still not clear if the SQD leads to a more efficient sampling of the wavefunction w.r.t the classical approach."
The recent IBM-Riken's research on the deceptive hybrid SQD methods published in Science ( this-last-paragraph ) for calculating some molecules such as N2 heavily relied on classical computers calculating the energies from artificially-chosen trial wavefunctions with No quantum computation nor quantum mechanical prediction
It is far better to use experimental values such as each atomic shape and properties from the beginning than to waste too much time in the meaningless time-consuming quantum mechanical calculations that cannot predict anything.
But the current unreal quantum mechanical shapeless atomic model based on unphysical antisymmetric wavefunctions prevents us from treating each molecule as a real object with shape and hampers developing useful multi-probe atomic force microscopes.
IBM founded in 1911 is clearly one of main culprits of protecting the unrealistic impractical quantum mechanics by spreading overhyped quantum computer fake news for investment fraud, cooperating with academia.
Unlike other overhyping social media companies such as Google and Microscoft, the older IBM with many Nobel prizes has technology of manufacturing ( quantum ) computers that has been craved by academia, universities that lack the latest computer technology.
So all the academia, universities needed to cooperate with IBM with the computer technology to make unreal quantum mechanics or computers appear to be promising for future (fake) technology to collect investment and taxpayers money by manipulating all the media by exploiting the current fake mainstream science power.
Actually, IBM is involved in the useless overhyped quantum computer's Shor's algorithm that cannot factor even simple 21, and a main culprit of intentionally blocking developing useful atomic force microscopes with multiple probes clarifying real atomic structures, selling their souls for the investment scam based on the current overhyped impractical quantum mechanics.

Feel free to link to this site.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}