Home page
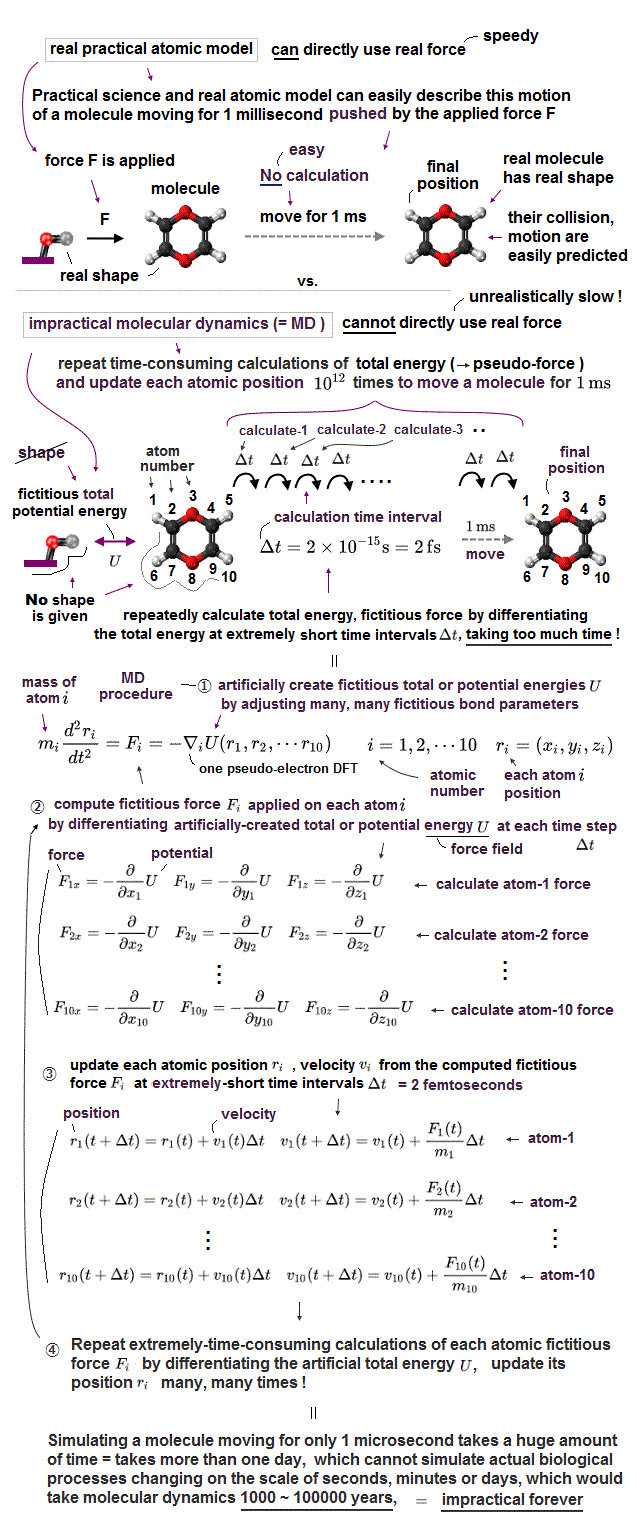
Today's overhyped useless physics
(Fig.1) Today's AI, Alphafold trained on today's bad experimental data cannot predict proteins, because the unreal quantum mechanics hampers developing useful multi-probe atomic force microscopes directly seeing atoms.
Today's medical research is deadend, unable to cure diseases such as cancers, Alzheimer.. because the present medical or biological researches do Not consider or clarify atomic mechanism of proteins or diseases, because the present unreal quantum mechanics, which cannot predict anything, hampers developing useful multi-probe atomic force microscopes directly seeing individual atoms .
Optical microscopes often used in today's medical research cannot see atoms (= each atomic size is 0.1nm = 1Å ) due to their bad resolution (> 100nm = 0.1μm, this-3rd-paragaph ).
↑ Even if they attach different fluorescent markers to different molecules or antibodies, this fluorescent microscope's resolution is too bad to see single atoms (= optical fluorescent microscopes can only vaguely detect light emitted from fluorescent markers instead of seeing atoms or proteins attached to the fluorescent markers ).
Today's best methods for seeing atomic structures of proteins are X-ray crystallography (= imaging proteins' structures by seeing X-ray diffraction or interference scattered by the target orderly-crystallized proteins ), NMR and cryo-electron microscopes (= cryo-EM ).
X-ray-crystallography, which is most often used in protein structure data band (= PDB ), is useless, unable to clarify atomic mechanism, because most proteins cannot be crystallized (= cannot be observed by X-ray crystallography ).
This-7th-paragraph says -- X-ray crystallography fail
"But a protein that never settles into a single form cannot be crystallized. The method that solved the structure of DNA and thousands of well-folded proteins is, for IDRs (= intrinsically-disordered region ), useless."
This-6th-paragarph says -- Useless X-ray
"the technique (= X-ray crystallography ) is not ideal for studying large, dynamic, or membrane-associated molecules."
This or this-1. crystalline sample says -- cannot crystallize
"The requirement for a crystalline sample is one of the most significant restrictions of x-ray crystallography....
Complex, big, or membrane-embedded proteins frequently cause them to fail, ( this-2nd-paragarph )"
This-5th-paragraph says -- Difficult crystallization
"membrane proteins,.. their structural characterization has been limited by challenges in stabilization and crystallization."
This-p.2-right-3rd-paragraph says -- Bad X-ray
"X-ray crystallography has its limitations, particularly
when studying large, flexible, or membrane-bound macromolecules
that are difficult to crystallize"
And even the luckily-crystallized proteins have unreal structures different from actual dynamical structures inside cells or bodies, and have bad resolutions. ← cannot be used for drug discovery.
This or this-X-ray crystallography-4th-paragraph says -- X-ray failed
"Large, complex molecules are often difficult to crystallize, especially when they contain dynamic areas.... Finally, the crystallization process can affect the molecular structure in a manner that means the final elucidated structure could be completely different to the native one found under physiological conditions"
NMR (= nuclear magnetic resonance vaguely measuring nuclear magnetic energy irrelevant to quantum mechanics ) is also useless, able to see only small molecules that must be purified ( this-Disadvantage of NMR, this-p.2-4th-paragraph, this-NMR-disadvantage ).
(Fig.C) Today's best microscope = Cryo-electron microscope cannot clarify atomic mechanism. → No cure for diseases, AI is useless.
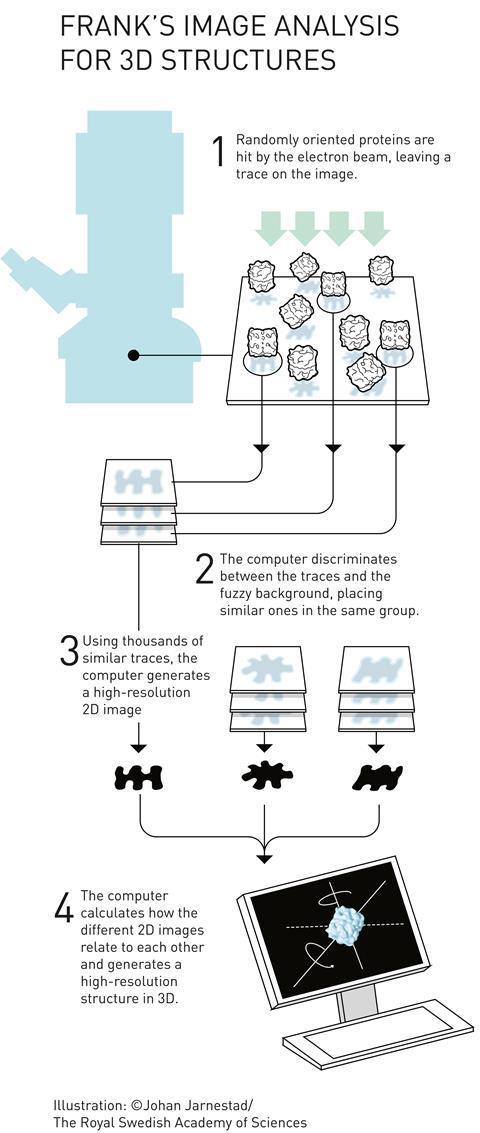
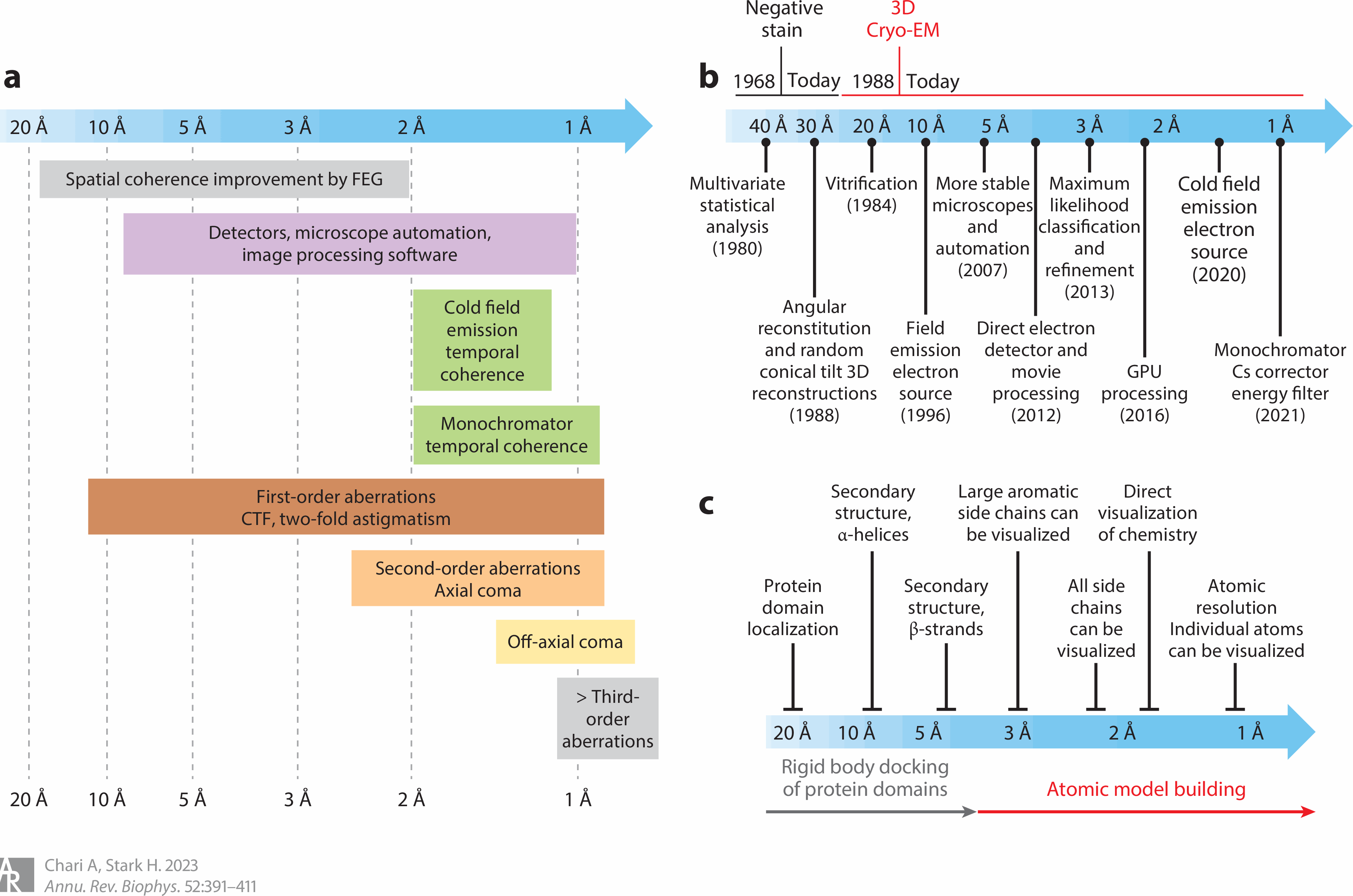
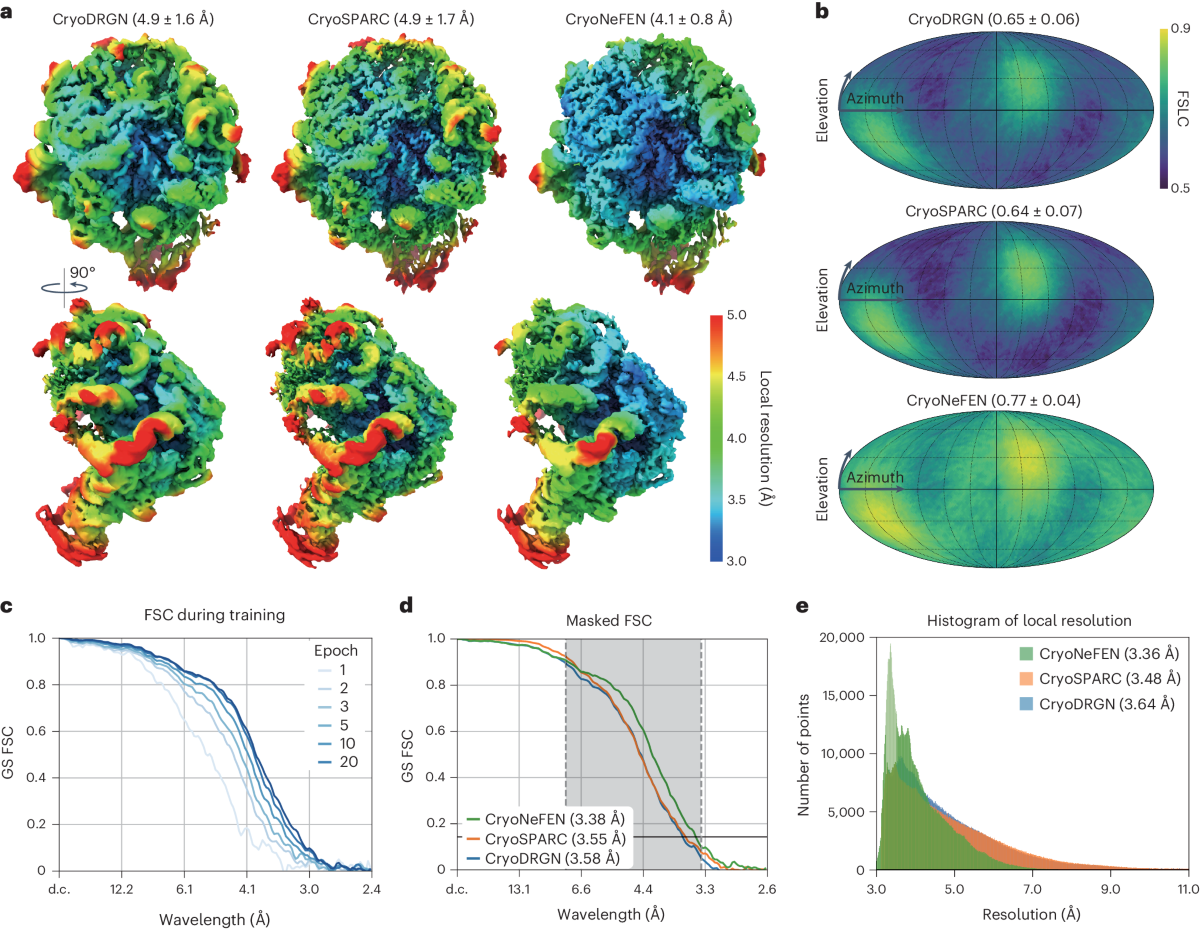
Cryo-electron microscopes (= cryo-EM ) seeing electrons randomly scattered by target proteins to image vague average protein structures are also useless with bad resolution that can Not see nor clarify atomic mechanisms ( this-p.4-5, this-cryo-EM-disadvantage ).
↑ In the sample of cryo-EM, the target (dirty) proteins often attached to various irrelevant molecules are placed and oriented randomly, which makes it impossible for the electron microscope to clearly see atomic structures of the target proteins ( this-2nd-paragraph~ ).
This-middle-Reconstruction-1st, 5~6th-paragraphs says -- Uncertain images
"When we covered our grid with a purified solution of proteins, we don't naively expect them to order themselves in any particular direction. Their orientation will, usually, be random ! "
"Before we get to actual 3D reconstruction, we need to visually isolate the good proteins. Remember, the noise-signal ratio in these micrographics is absolutely awful; many of the proteins here will be degraded,.. the micrographics may have contaminates,"
This or this-introduction-2nd-paragraph says -- Bad cryo-EM's image
"However, inferring information about biomolecular conformational heterogeneity from cryo-EM images is challenging"
This-9th-paragraph says -- Cryo-EM's blur image
"cryo-EM works by averaging thousands of images of the same molecule to reconstruct a 3D model. When the protein being studied is in constant motion, shifting between dozens or even hundreds of conformations, the resulting image is often a blurry approximation"
This-p.11-conclusions say -- Cryo-EM's bad resolution
"Although the current highest resolution
reached 1.2 Å, most of the reported cryo-EM structures
are still at the 3–4Å level (= cryo-EM cannot see single atoms of 1Å size, this-p.9 ), and therefore the structural
information on drug molecule binding target sites usually needs to be combined with higher resolution protein
structures obtained from X-ray crystallography"
This-7~8th-paragraphs (2025) say -- cannot see atoms
"However, cryo-EM's resolution typically doesn't match the atomic-level precision of crystallography"
This or this-Limitations of cryo-EM (2024) says -- Useless cryo-EM
"There are resolution limitations and sometimes structural details cannot be well-resolved.
Larger samples are difficult to study."
Even in the latest researches in 2025, cryo-EM cannot get the atomic level resolution (< 1Å = angstrom ), as shown in this-abstract-5.8Å resolution, this-p.5-6Å resolution this-p.1-3.04Å resolution, this-p.3-2.8Å resolution, this-p.3-5-3.75Å resolution, this-12Å resolution, this-p.5d-red-8.5Å.
↑ Cryo-electron microscope's resolution of membrane proteins is too bad ( this-p.6-left-last-paragraph says resolution of 23.4 ~ 33.0 Å this-p.2-right-middle-local resolution-blue ~ 16.0 Å ← far worse than atomic resolution of 1Å this-middle-Data resolution )
This-last-Summary and outlook (2023) says -- No atomic resolution
"breaking the 1Å resolution barrier in single-particle cryo-EM should Not be expected to happen routinely."
In cryo-electron microscopes, it is extremely difficult to purify homogenious proteins (= isolating only some target proteins means protein-protein interactions or enzymatic reactions cannot be clarified by cryo-EM ) = if target proteins' conformations are flexibly changed or mixed with irrelevant molecules, cryo-EM cannot precisely determine the atomic structures of proteins ( this-lower-Challenges and limitation, this-3rd~4th-paragraphs ).
This (or this )-middle-current limitations and challenges in cryo-EM say
"Sample heterogeneity: Membrane proteins can be difficult to purify and can exist in multiple conformational states."
As a result, in X-ray crystallography and cryo-electron microscopy (= EM ), it is impossible to know the precise ( native ) atomic structures especially in flexibly-changing area (= which flexible molecular area is the most important for enzymatic reactions ) that are blurred and uncertain after averaging measured protein data (= in cryo-EM, positions of each protein are random, uncertain, which can never get the precise atomic structures of proteins. )
This-p.3-left-1st-paragraph-(b) says -- Flexible protein difficult
"the requirement of
sample homogeneity is very high, it will be difficult to get
good results if the protein is flexible"
This-p.1-right-last-paragraph says -- No RNA
"The scarcity of protein-free RNA cryo-EM structures... intrinsic heterogeneity in most RNA molecules
greatly limits the attainable resolution by SPA (= cryo-EM )"
This-p.4-left-last-paragraph (2025) says -- No sugars
"Although the current resolution
does Not allow for precise assignment of the sugar moieties (by cryo-EM)"
This-abstract-last (2024) says -- cryo-EM fail
"Despite their inherent importance, accurately depicting PTMs (= post-translational-modifications ) in experimental
studies of protein structures often poses a challenge"
(Fig.P) Why today's AI, Alphafold cannot develop drugs ?

Today's microscopes unable to clarify native atomic structures is why the overhyped AI, Alphafold that are trained only on proteins structures in PDB ( this-p.2-right ) obtained by these useless X-ray crystallography, NMR, cryo-EM (= bad, non-atomic resolution ) cannot predict real atomic structures of various proteins nor develop effective drugs.
This-1st paragraph(6/25/2025) say -- AI useless
"AI won't revolutionize drug discovery"
This-3rd-paragraph (9/10/2025) says -- No drug discovery
"yet there are few AI-discovered candidates in late-stage clinical trials, and not one has been approved ( this-abstract )"
This-p.3-right-last~p.4-left (2024) says Useless Alphafold
"It is still an open question whether the structures in a cellular environment can be truthfully reproduced by
AlphaFold2 prediction or in in vitro experiments"
This-7~8th, 10th paragraphs (2025) say -- lack protein data
"AI oversimplifies the representation of the protein's flexible regions."
"while AI tools like AlphaFold are powerful, their training data lacks information about complex protein behavior"
This-middle-cryo-electron microscopy-2nd, 3rd, 7th~ paragraphs (2025) say
"However, cryo-EM is not without its limitations... The images produced are averages of thousands of molecular snapshots, meaning that for highly flexible proteins, the final structure can be blurry or incomplete."
"X-ray crystallography requires proteins to be coaxed into a crystalline form,.. not all proteins crystallize well. "
"The core issue AI isn't just missing data — AlphaFold's entire approach is built on assumptions that don't apply to disordered proteins (= related to diseases, so AI cannot cure diseases )"
The 8th-paragraph of this news (10/29/2025) says -- AI wrong
"In more than half of the cases, the models predicted the structure as if the interferences in the amino acid sequence had never occurred. This shows us that even the most advanced AI models do Not really understand why a drug binds to a protein; they only recognize patterns that they have seen before,"
(Fig.A) Overhyped fake news on seeing atomic mechanisms of cells is rampant in medical research. ↓
The 2nd-last paragraph of this hyped news on the alleged atomic level DNA transcription (2025) says
"Our findings may (= uncertain speculation, so still useless ) contribute to a better understanding of diseases"
↑ This research used cryo-EM whose resolution was very bad (= 3.0Å ~ 15Å, this-p.10-left, this-p.6~8 compared with this and this ), which cannot distinguish single atoms of 1Å size, hence No atomic mechanism was clarified, contrary to hypes.
This other hyped research on the alleged molecular mechanism of brain blood ( this paper ↓ )
p.4-Figure 1G's transmission electron microscope's picture cannot see nor distinguish single atoms (= so No atomic mechanism was clarified ).
p.12-Limitaions of the study-right-middle says -- No atoms seen
"Moreover, the
regional specificity of N-cadherin expression in the brain remains
poorly understood" ← still atomic mechanism cannot be understood, contrary to hypes.
News claiming "clarifying atomic or molecular mechanisms" is fake and hyped.
For example, this-p.20-right-Limitations of the modeling approach says
"These are based,
respectively, on cryo-EM structures.. and X-ray structures
of prokaryotic sodium channel.... Since the experimental structures
are obtained in lack of lipid membranes and membrane voltage,
some of their features may be non-native (= unreal structures ).... Furthermore, in lack of experimental
structural data on the C-terminal part of linker I/II we refrained
from an attempt to de novo model this part"
This-lower-Next step says -- Unclear atomic mechanism
"It is likely that the conformational changes we observe are controlled by yet-to-be-found regulators in cells" ← still atomic mechanisms are unclear.

Feel free to link to this site.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}