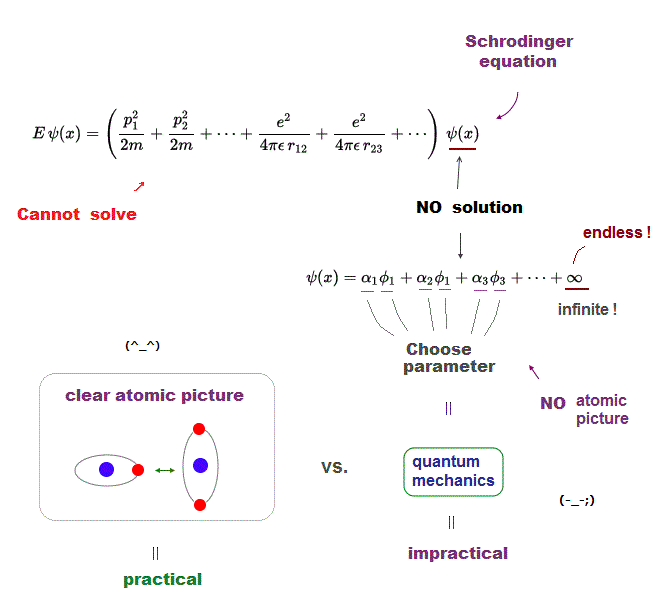
(Fig.1) Medical research cannot clarify atomic mechanism nor cure diseases due to bad electron microscopes that cannot see atoms.
The present medical research (and hyped AI ) has been deadend with No progress (= still No cures for cancers or Alzheimer ).
Because the best mainstream method for clarifying molecular mechanisms in medical research has been (useless) hyped cryo-electron microscope (= cryo-EM ), which can Not clarify atomic mechanism due to its bad resolution ( this-limitations of cryo EM ), since 1970 (= No progress for long years, this-history ).
Only multi-probe atomic force microscopes (= newer than the old electron microscopy ) can directly see and clarify atomic mechanism of proteins for curing diseases ( this-p.3-right-atomic resolution imaging ), which has been hampered by the unreal quantum mechanical atomic model for a long time.
The 2nd, 3rd paragraphs of this hyped news (8/21/2025) say
"a team of researchers.. has succeeded in establishing the first comprehensive transport model of the plasma membrane Ca2+-ATPase (PMCA) by resolving its 3D structure" ← fake news
"This mechanism could be a promising (= still useless ) starting point for developing new drugs"
↑ This research paper ↓
p.2-left-2nd-paragraph says "we determined an ensemble of PMCA2 (= plasma membrane Ca2+-ATPase ) structures.. by cryo-electron microscopy (cryo-EM)"
p.2-right-2nd-paragraph says
"The eight resulting structures of PMCA2 had an overall resolution
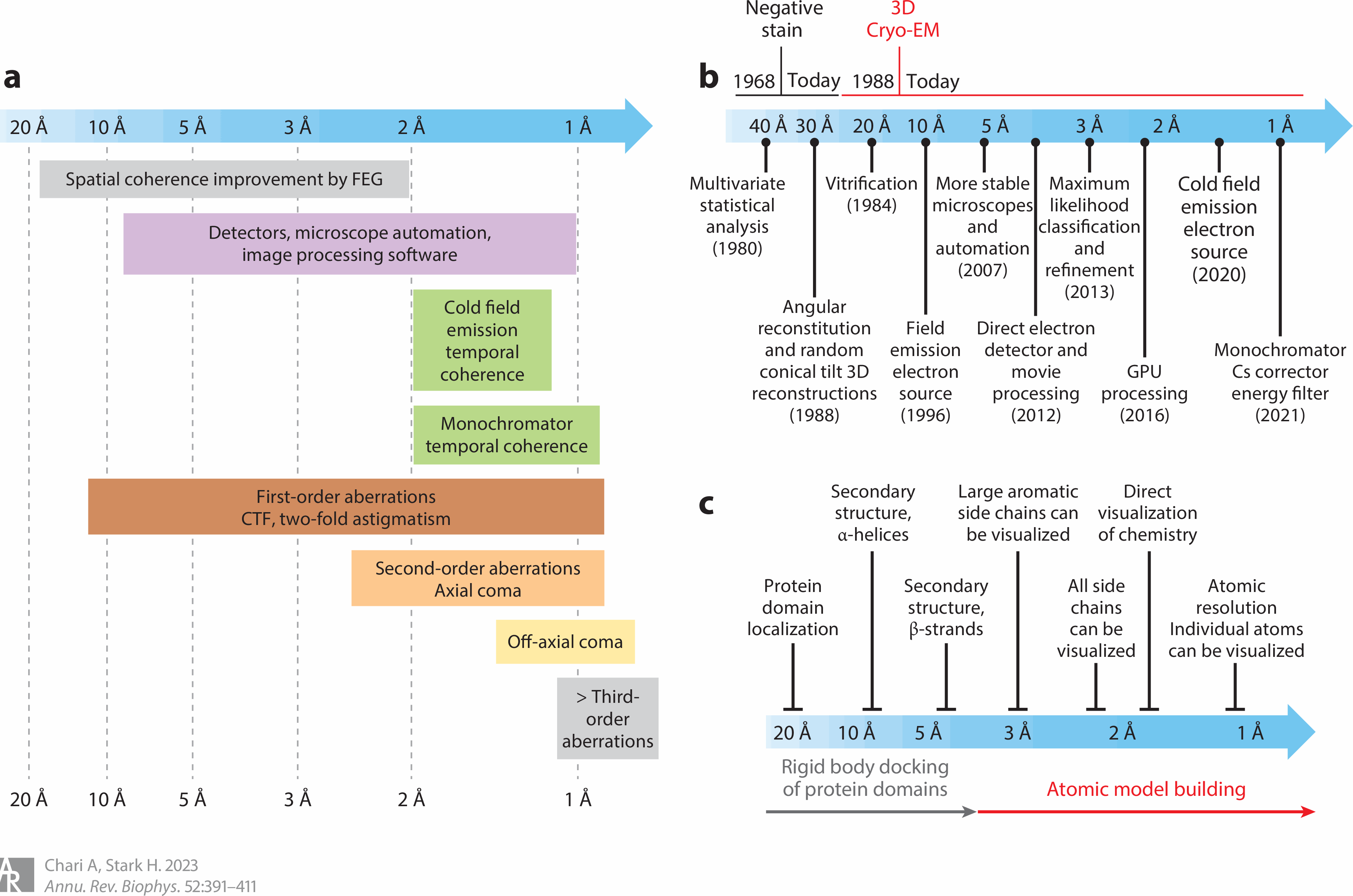
ranging from 2.8 Å to 3.6 Å. ← Cryo-electron microscopes are useless, unable to get atomic resolution of less than 1Å (= each atomic size is less than 1 angstrom or 1Å, this-middle-picture's cryo-EM's resolution is worse than the atomic 1Å = 1 Angstrom resolution )
p.13-right-1st-paragraph says "Because the local resolution of Ig1 was still too low to allow for de novo modelling"
p.25-even this latest Cryo-EM's (map) resolution was too bad (= 6.3Å, 9.5Å, this-p.3-p.6-local resolution ), which cannot clarify atomic mechanism (of proteins or diseases ) needing less than 1Å resolution, so cannot cure cancers or Alzheimer.
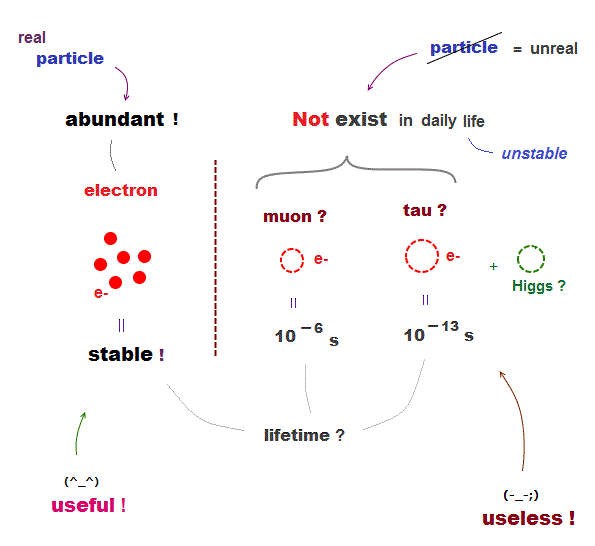
The 4th~7th, 4th-last paragraphs of this hyped news (8/27/2025) say
"high-resolution cryo-electron microscope (= cryo-EM )... was used in the current study to elucidate the static structure of a molecular machine"
"These structures are very important, but Not sufficient.... we need to understand how they move.... This is a task that is even more challenging" ← Today's best cryogenic microscopes are useless with bad resolution, and unable to clarify dynamical proteins' motion
"NMR data is often very abstract." ← NMR is also useless ( this-disadvantages of NMR ).
"Molecular dynamics (MD) simulations are used to calculate dynamic structural models that visualize the structural changes. However, these models require experimental verification." ← MD is useless, too time-consuming, unable to predict anything due to artificially-chosen force-field pseudo-potential that must be fitted to experimental results ( this-p.5-lower-Limitaions of MD ).
"The researchers have Not yet been able to prove a direct connection," ← atomic mechanism of proteins could Not be clarified after all.
↑ This research paper ↓
p.3-left-3rd-paragraph-Results and discussion says
"We determined the structure of Exo9 (= RNA degrading proteins ).. by X-ray crystallography to 3.8 Å resolution
and by cryo-EM to
3.2 Å resolution ( this-p.37-resolution range )" ← Today's methods can Not clarify atomic mechanism, which needs much better < 1Å (= 1 angstrom ) resolution.
p.4-left-last-paragraph says "Rrp42-EL, that is unresolved and thus invisible in both the X-ray and cryo-EM structure" ← There are still many proteins that cannot bee seen even by today's best X-ray crystallography or cryo-EM.
p.5-Fig.3-last says "Rrp42-EL is Not visible in the structure (= still invisible proteins ). For clarity only the outline of the subunit that is NMR active (= dots ) is shown" ← Most protein subunits cannot be seen by NMR.
p.7-Fig.5B shows the time consuming MD could simulate only 100ns of the target protein, which is useless, cannot simulate much-longer proteins' motions.
↑ Thic current only molecular-simulating method = molecular dynamics (= MD ) is based on artificial potentials called force fields (= which prediction often fail ) whose interaction parameters must be experimentally adjusted (= quantum mechanical DFT also needs artificially-chosen exchange pseudo-potentials that depend on experimental values with No ability to predict anything ).
↑ But None of today's experimental methods such as X-ray crystallography, cryo-electron microscopy, NMR can get precise atomic structures of proteins, so MD (and DFT ) simulation needing experimentally-adjusted parameters of its force-field potential energies is also useless.
↑ Even if the MD's force field's potential parameters are right, the precise initial protein structures are unavailable in the current bad microscopes (= proteins of the same amino acid sequences can take many unknown different 3-D structures ), which makes MD dynamical simulaion give wrong protein structure results.
This-p.3-1st,2nd,3rd-paragraphs (7/2025) say
"traditional force fields (of MD ) were largely developed and parameterized to describe well-folded proteins, often leading to over-compact ensembles for disordered systems"
"it remains unclear which force fields
best describe peptides across different structural regimes"
"While No force field performs optimally in all cases"
↑ It is impossible for today's MD based on artificial force field potential (and useless quantum mechanics ) to predict precise proteins' motions.
Only multi-probe atomic force microscopes can directly clarify atomic mechanisms of proteins and diseases, which is hampered by unreal quantum mechanical model now.

Feel free to link to this site.
{kind=link}
{kind=link}
{kind=link}
{kind=link}