
(Fig.1) If we try to make the three-electron wavefunction mixing the up and down spins, this wavefunction vanishes by canceling the plus and minus terms due to the paradoxical quantum mechanical Pauli antisymmetric rule !
Textbooks often write about two-electron helium and hydrogen molecule, but rarely mention three-electron atoms such as Lithium or molecules.
Because quantum mechanical Pauli principle is unable to treat all atoms and molecules with more than two electrons mixing the up and down or spacial symmetric and antisymmetric wavefunctions ( this p.3-6, or this-(1)-(11) ) !
In the upper figure, the atom-A has an electron with down-spin (= down-spin ↓ is often denoted as β ), the atom-B has an electron with up-spin (= up-spin ↑ is often denoted as α ), and the atom-C has an electron with up-spin.
So the molecular bond attractive (= positive ) exchange energy based on spatial symmetric wavefunctions (= singlet ) between two up-down anti-parallel spins must be generated between an electrons-1 in A wavefunction (= down-spin ) and an electron-2 in B wavefunction (= up-spin ), and between electrons-1 in A wavefunction (= down-spin ) and electron-3 in C wavefunction (= up-spin ).
See this p.2, this p.1-4, this p.8, this p.22
And the Pauli repulsive (= negative ) exchange energy based on spacial antisymmetric wavefunction (= triplet ) between the same up-up spins must be generated between electrons-2 in B wavefunction (= up-spin ) and electron-3 in C wavefunction (= up-spin ).
↑ These spatial symmetric and antisymmetric wavefunctions have different inconsistent normalization coefficients, so all these separate wavefunctions must be united into one wavefunction mixing spacial symmetric and antisymmetric wavefunction to explain atoms and molecules with more than two electrons, though this is impossible in the quantum mechanical Pauli antisymmetric wavefunctions.
For example, we prepare one term which is the product of three atomic wavefunctions of φA↓(1) (= atomic-A wavefunction with down spin containing the electron-1 ), φB↑(2) (= atomic-B wavefunction with up spin containing the electron-2 ), and φC↑(3) (= atomic-C wavefunction with up spin containing the electron-3 ) = φA↓(1)φB↑(2)φC↑(3).
When exchanging two electrons 1 and 2, this term's sign should remain unchanged as "positive", and this term's electrons' labels change into φA↓(2)φB↑(1)φC↑(3), because the electron-1 exists in the atomic-A wavefunction with down spin and the electron-2 exists in the atomic-B wavefunction with up spin, which must form the spatial symmetric wavefunction (= singlet ) with respect to interchanging two electrons 1 and 2 in these terms.
Next we exchange the electron 2 and 3 in this second term of φA↓(2)φB↑(1)φC↑(3), which changes into φA↓(3)φB↑(1)φC↑(2)
↑ No sign change of these spatial symmetric wavefunctions, because the electrons-2 and 3 exist in the atomic-A with down spin and the atomic-C with up-spin. = the up-down spin wavefunction must be symmetric (= singlet ) which doesn't change the sign under the exchange of two electrons.
In the same way, when we exchange two electrons 2 and 3 in the first term of φA↓(1)φB↑(2)φC↑(3), this term flips its sign like -φA↓(1)φB↑(3)φC↑(2).
↑ Because the electrons-2 and 3 exist in the atomic-B wavefunction with up-spin and the atomic-C wavefunction with the same parallel up-spin, hence, these two electrons with the same up-up spin must form the spacial antisymmetric wavefunction (= triplet ) which must change its sign under the exchange of two electrons' coordinates or labels.
And if we exchange two electrons 1 and 3 in this term of -φA↓(1)φB↑(3)φC↑(2), this does not change the sign like -φA↓(3)φB↑(1)φC↑(2).
↑ because the electrons 1 and 3 exist in the atomic-A with down spin and the atomic-B with up spin, which must form the spatial symmetric wavefunction (= singlet ) in the term of -φA↓(1)φB↑(3)φC↑(2).
As a result, this three-electron molecule needs the contradictory terms of plus (= φA↓(3)φB↑(1)φC↑(2) ) and minus ( -φA↓(3)φB↑(1)φC↑(2) ) signs, which cancel each other !
↑ This result shows the quantum mechanical Pauli principle cannot be applied to the case of three electrons or more than two electrons mixing up and down spins.
Only the spatial wavefunction (= not spin part ) is important, contributing to the total energy, but the contradictory quantum mechanical singlet-triplet rule cannot obtain the legitimate spacial wavefunction for three-electron Lithium separated from spin part ( this p.12-third-paragarph ).
For example, the three-electron Lithium (Hylleraas) wavefunction ( this p.3-(6) ) must contain the paradoxical terms like the exchanging the 1st and 3rd electrons (= exchanging the 1st and 3rd wavefunctions ) paradoxically changes the sign φ(1,2,3) → -φ(3,2,1) (= 1st-3rd-electrons-1 and 3 exchanged ) or does Not change the sign -φ(2,3,1) → -φ(1,3,2) (= 1st-3rd-electrons-1 and 2 exchanged ), which is inconsistent with respect to symmetric or antisymmetric wavefunctions under exchanging two electrons, and disagrees with the original Pauli antisymmetric rule.
(Fig.2) In Pauli antisymmetric wavefunction or Slater determinant, only Pauli repulsive exchange energy with the same spins or triplet remains. Any singlet exchange energies (= molecular attractive bond ) with the up-down spins are zero.
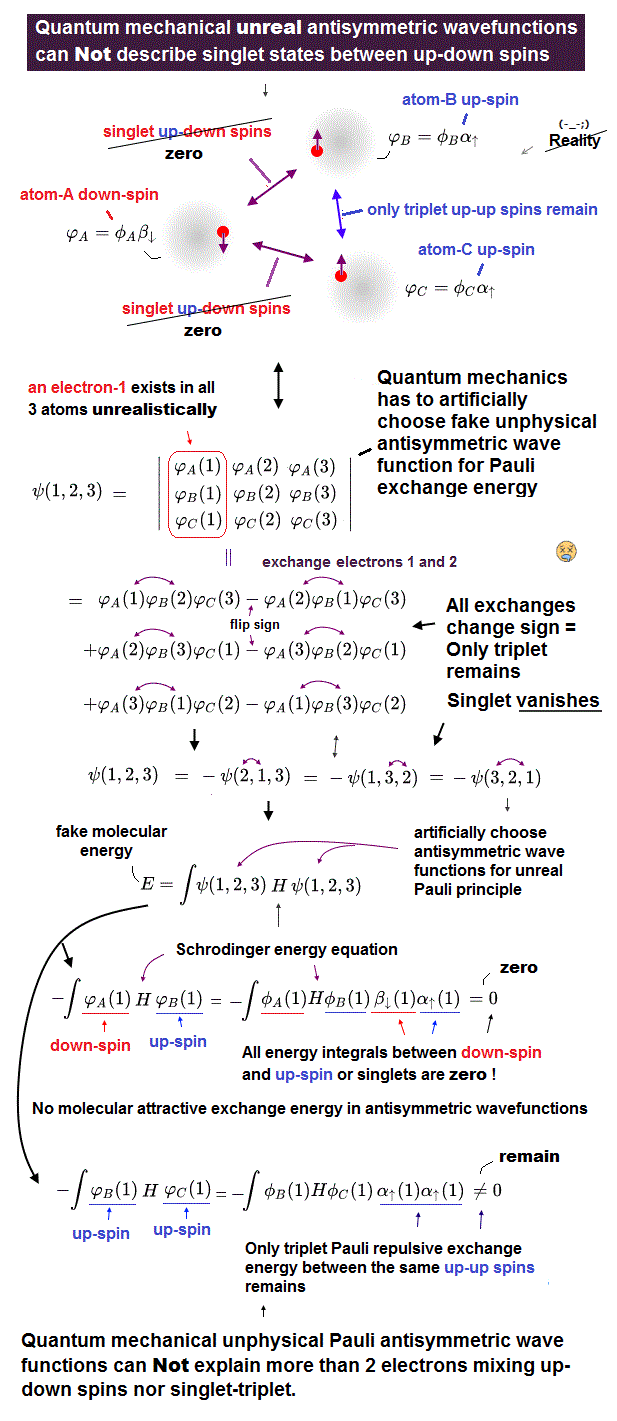
Quantum mechanical Pauli principle cannot describe atoms or molecules with more than two electrons mixing singlet and triplet spins, which means quantum mechanics cannot describe the multi-electron wavefunction mixing spacial symmetric wavefunction (= causing molecular bond attractive exchange energy between up-down singlet spins, this p.3 ) and antisymmetric wavefunction (= causing Pauli repulsive antibond exchange energy between up-up or down-down triplet spins ).
To deal with this serious paradox, quantum mechanics artificially changed the rule, and started to say all multi-electron atomic and molecular wavefunctions must be expressed only as (nonphysical Pauli) antisymmetric wavefunctions (= No symmetric spacial wavefunctions ) or Slater determinant ( this p.1-5 ) where No molecular bonds expressed as attractive symmetric exchange energies (or No bonding exchange integral ) can be generated when atoms or molecules contain more than two electrons. ← No molecular bonds ! It means quantum mechanics is false.
In quantum mechanics, all wavefunctions must be expressed as Pauli antisymmetric wavefunctions or Slater determinants ( this p.7-8 ) where each atomic wavefunction consists of spatial wavefunction and spin wavefunction parts (= α↑ is up-spin, and β↓ is down-spin ).
In exchange integrals of two different atomic wavefunctions, the spin integral of two different up (= α↑ ) and down (= β↓ ) spins always becomes zero (= ∫αβ = 0 = No molecular bond attractive exchange energy between the different up-down spins ), and only the spin-integral of two same up-up or down-down spins remains as non-zero (= only Pauli repulsive antibonds between the same up-up or down-down spins remain ) in atoms or molecules with more than two electrons ( this p.4-spin-selection-rule ).
↑ It means in quantum mechanics, No molecular attractive exchange energy integral between two different up-down spins is generated (= No molecular bonds in quantum mechanics ! ), and only unnecessary Pauli repulsive exchange energy between the same up-up or down-down spins remains ( this p.5-6, this p.37, this p.10-lower ).
This is a fatal flaw of quantum mechanical Pauli principle based on unphysical antisymmetric wavefunctions.
(Fig.3) ↓ Singlet-triplet theory is contradictory in Pauli principle of more than two electrons, so quantum mechanics is false.

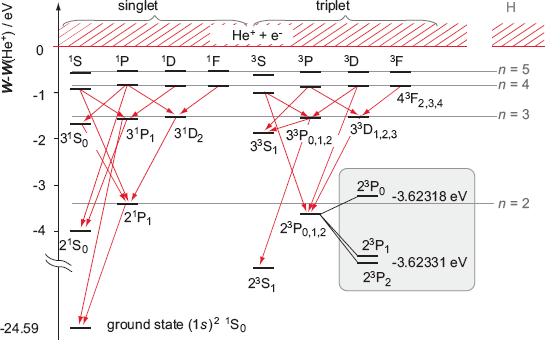
In two-electron helium atom, quantum mechanics insists there is difference between the energy levels (= ex. 1s2p of helium ) of singlet (= 1P ) expressed as spin-up ↑ (= α ) and spin-down ↓ (= β ) and triplet (= 3P = spin-up-up, spin-up-down, spin-down-down symmetric ) wavefunctions ( this p.9 ).
But as shown in this, this quantum mechanical singlet-triplet theory is invalid in more than two electrons such as lithium.
So quantum mechanics illegitimately changed the original Pauli principle definition based on the helium-singlet-triplet states into generalized atomic Pauli principle version expressed as Slater determinant or Pauli antisymmetric wavefunction where each wavefunction consists of spatial (= φ ) and spin (= α-up-spin, β-down-spin ) parts (= which contains No spatial symmetric wavefunction even in spin-up-down ↑↓ case ).
The problem is this generalized Pauli principle antisymmetric wavefunction or Slater determinant can Not describe nor distinguish singlet and triplet energy levels. = There is only one form of antisymmetric wavefunction (= two terms connected by minus ) with two electrons of spin-up and down, as shown in the above figure.
Some people say combining two Slater determinants by exchanging spins between two wavefunctions can distinguish these singlet and triplet states ( this p.4-9 ).
↑ But this method based on swapping spins between two atomic wavefunctions cannot be used in cases of more than two electrons.
So in all atoms with more than two electrons, the original singlet-triplet theory ( this p.8 ) is invalid, which is inconsistent with the quantum mechanical claim that even magnesium and calcium with more than two electrons should have singlet and triplet energy levels.
This clear self-contradiction shows quantum mechanics is wrong.
(Fig.3') ↓ Beryllium (= Be ) and Boron ion (= B+ ) having exactly the same electrons have completely different orders of energy levels. ← No consistent rules in quantum mechanics.
Quantum mechanical Schrodinger equations are unsolvable, unable to predict any atomic (ground and excited-state) energy levels.
Furthermore, even quantum mechanical artificial rules are self-contradictory, and unphysical.
Beryllium (= Be ) and singly-ionized boron (= B+ ) have exactly the same structure with two valence electrons, so these two atoms should have the same-order energy levels.
But the orders of 2s3s and 2p2 between Be and B+ are completely different and inconsistent.
Boron (= B ) and singly-ionized carbon (= C+ ) also have exactly the same electron structure with the three same valence electrons, but the energy positions of 2s23s are completely different between B and C+, which is contradictory.
Helium's three energy levels of 1s2p triplet (= 3P = 2,1,0 ) are inconsistent with beryllium's (= 2s2p ) and magnesium's (= 3s3p ) triplet states' order 3P = 0,1,2. ← No legitimate consistent explanation in quantum mechanics.
In boron with three valence electrons, the 2s-2p2 state splits into five different energy levels (= 4P + 2D ), while in aluminum that also has three valence electrons, the same type 3s-3p2 state splits into only three energy levels (= 4P ), which is inconsistent (= Aluminum paradoxically ignores 3p2 singlet inside 3s-3p2 state ). ← Singlet-triplet based on the original Pauli principle becomes invalid in three valence electrons.
In carbon with four valence electrons, 2s2-2p2 splits into five states (= probably they assume three triplets + two singlets in 2p2 inside 2s2-2p2, ) while the same type 2s2-2p3p paradoxically splits into ten states (= only this includes two types of triplets ), though 2s2-2p2 and 2s2-2p3p should have the same types of triplet.
In neon with eight valence electrons where they probably try to treat the 6 electrons as one independent singlet state with spin-zero, then, they should split the remaining 2p-3s (of 2p53s ) into three triplet + one singlet like helium and beryllium, but they split 2p53s into two pairs irrelevant to the original triplet-singlet theory, which is inconsistent.
(Fig.4) Molecular orbital (= MO ) theory requires each single electron to exist in multiple different atoms simultaneously, which also causes unrealistically-inseparable electrons or atoms, and MO cannot avoid unnecessary Pauli repulsive exchange energy, so false.
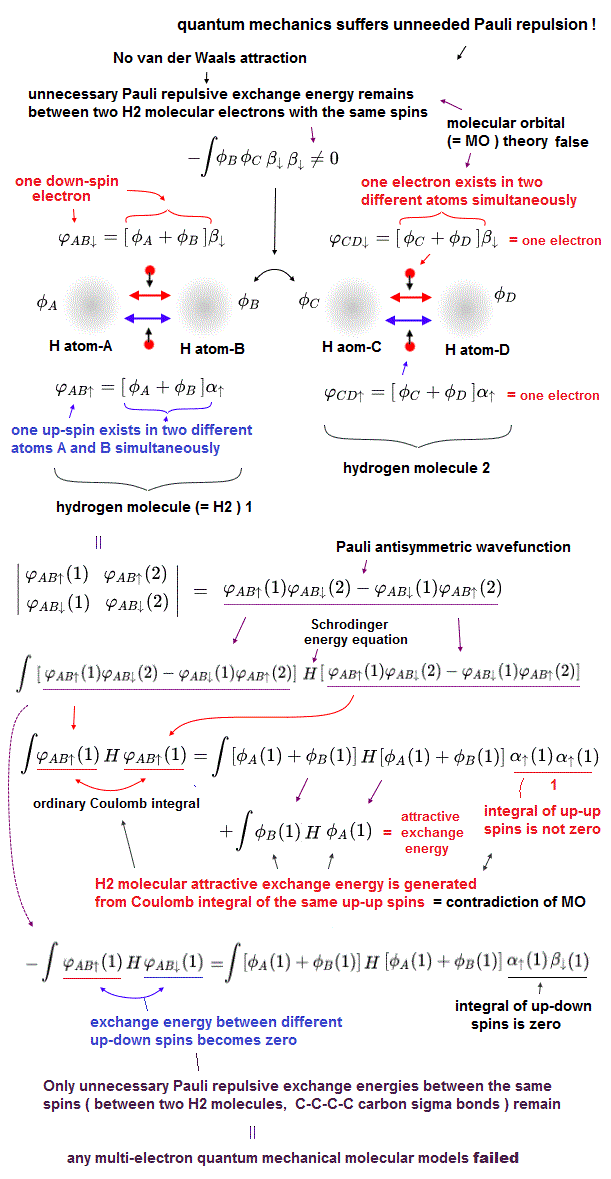
Quantum mechanical Pauli principle expressed as antisymmetric wavefunction or Slater determinants have fatal flaws, and it can Not describe any molecular bond attractive exchange energies between the different up-down spins, and only unnecessary Pauli repulsive exchange energies between the same up-up or down-down spins remain.
To handle this quantum mechanical contradiction, the molecular orbital theory (= MO ) was invented, though this ad-hoc molecular orbital theory cannot eliminate unnecessary Pauli repulsion after all, either.
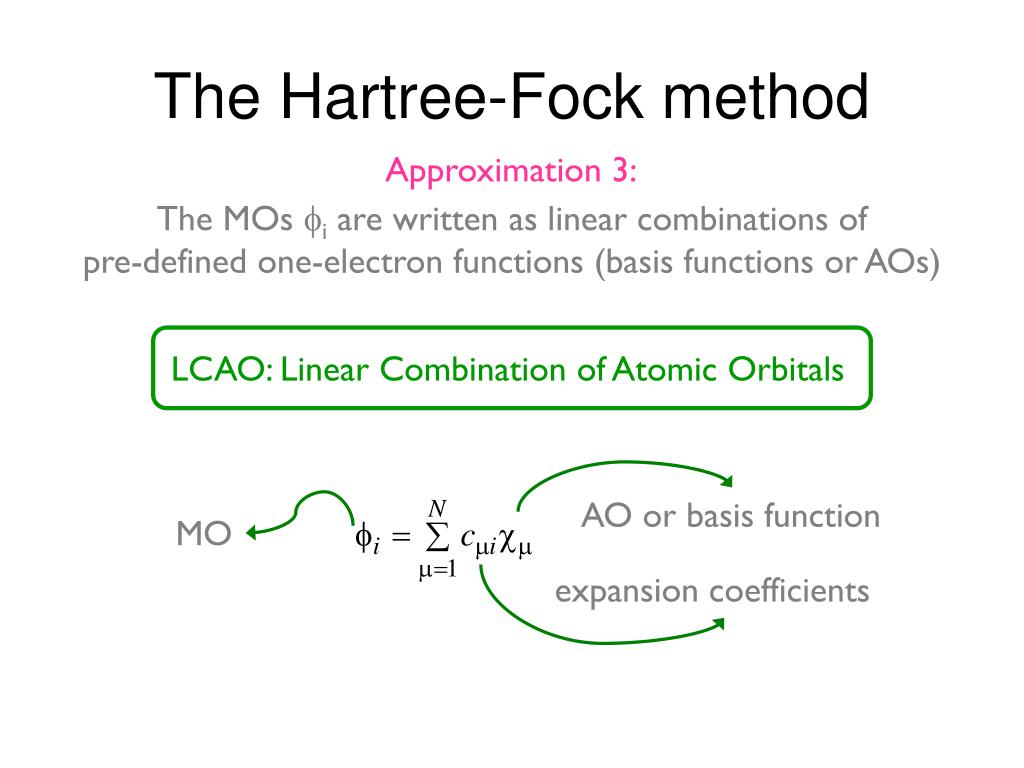
In this ad-hoc molecular orbital theory, each single electron must always spread over more than one atoms or orbitals = each electron's molecular orbital always becomes a linear combination of multiple atomic wavefunctions ( this p.7(or p.5), this p.4, this p.3 ). ← This unrealistic condition is necessary for generating the fictitious molecular bond attractive energies in MO by avoiding the vanishing attractive exchange energies between the up-down spins.
It means electrons of molecular orbital theory are unrealistically inseparable into different atoms due to their constantly-spreading molecular orbitals, and the ad-hoc molecular orbitals cause non-existent ionization states ( this p.27, this p.14-15-(11)-(13) ), hence, the molecular orbital theory often failed to explain experimental results ( this p.25 ).
This molecular orbital wavefunction also has to be expressed as the unphysical Pauli antisymmetric wavefunction or Slater determinant consisting of multiple molecular orbitals ( this p.14, this-(9.6.1)-(9.6.2) ) where each orbit consists of a spatial molecular orbital (= linear combination of multiple atomic orbitals ) and spin part.
↑ This means the molecular orbital theory also causes unnecessary Pauli repulsive exchange energy between two molecular orbitals with the same spins, which means any quantum mechanical multi-electron models failed to incorporate Pauli principle expressed as the antisymmetric wavefunctions or Slater determinants.
To avoid this unnecessary Pauli repulsive exchange energies, all molecular orbitals must be "orthogonal" to each other, which means all exchange energies or overlap integral between any two different molecular orbitals must be zero ( this p.6, this p.3 ).
↑ When any exchange energies (= or overlap integral ) between two different orbital wavefunctions become zero due to their orthogonal wavefunction's relationship regardless of their spin states, neither molecular bond attractive exchange energies nor annoying unneeded Pauli repulsive exchange energies seem to appear in the energy calculations. ← But even these artificial "orthogonal" wavefunctions cannot avoid unnecessary Pauli repulsive energies after all, so the molecular orbital theory is wrong, as I explain later.
If all exchange energy integrals become zero due to artificially choosing the orthogonal molecular orbital wavefunctions, how does the ad-hoc molecular orbital theory generate the molecular attractive (or Pauli repulsive ) exchange energies, though the exchange energy is zero ?
As shown in the upper figure, even if exchange energies or exchange integrals are zero due to the artificially-chosen orthogonal wavefunctions, the normal Coulomb integral parts (= the product of two same wavefunctions with the same spin ) automatically contain the molecular attractive exchange energies caused by each molecular orbital which is a linear combination of multiple atomic orbitals, which is the trick of molecular orbital theory to generate molecular attractive exchange energies.
Each molecular orbital = ψ = φA(1) + φB(1), hence, the normal Coulomb integral of ∫ |ψ|2 = ∫ | φA(1) |2 + | φB(1) |2 + ∫ 2 φA(1)φB(1) ← the attractive exchange energy +∫φA(1)φB(1) is automatically contained in the normal Coulomb integral of each single molecular orbital.
↑ Even this artificial molecular orbital manipulation cannot eliminate the unnecessary Pauli repulsion, so physicists rely on the impractical time-consuming configuration integral (= CI ) using unphysical negative exchange energy between two fictitious electrons in different Slater determinants to lower the total energy, which CI trick is ad-hoc and completely unrealistic.
Physicists gave up the theoretical prediction based on this failed molecular orbital theory or multi-electron quantum mechanical model, and instead, invented the empirical Huckel method which can treat only π bonds at first (= because p orbital originally consists of plus and minus phases, and their π bond molecular orbitals can easily become orthogonal ) by artificially adjusting free semi-empirical energy parameters obtained from experiments ( this p.4 ), which cannot predict any molecular enregies or treat spin exchange energies ( this p.2-2nd-last paragraph ).
As shown here, quantum mechanical Pauli principle expressed as unphysical antisymmetric wavefunctions or Slater determinants intrinsically contradict multi-electron atomic or molecular wavefunctions.
This is why only unrealistic one-pseudo-electron approximation called density functional theory (= DFT ) remains as the most-widely used quantum mechanical calculation tool, though this DFT also failed to incorporate Pauli principle.
(Fig.5) Helium (= He ) atom contains two electrons A, B with up and down spins. H atom contains one electron C with up spin. In this case, unnecessary Pauli repulsion occurs between He and H atoms due to zero singlet attractive exchange energy between A and C electrons.

As shown in this, quantum mechanical Pauli antisymmetric wavefunctions or Slater determinants can Not describe cases containing more than two electrons or mixing singlet-triplet spins.
We think about the case where one helium atom (= He ) contains two electrons A (= φA ) and B (= φB ) with up-down spins, and one hydrogen atom (= H ) contains one electron with up spin, as shown in upper figure.
In this case, we can make the singlet or attractive (= positive ) exchange energy between A and B electrons only inside He atom by combining two unphysical Slater determinants artificially swapping two up-down spins of A and B electrons of He atom.
↑ But even in this case of using multiple Slater determinants, we can Not express the singlet attractive exchange energy between A electron with down spin of He atom and C electron (= φC ) with up spin of H atom, so unnecessary Pauli repulsive exchange energy remains, which cannot describe van der Waals attraction between H and He atoms.
So quantum mechanical Pauli antisymmetric wavefunctions or Slater determinants are proved to be wrong.
As shown in the upper figure, we prepare two Slater determinants where one Slater determinant (= Slater-1 ) contains A electron with down-spin and B electron with up-spin inside He atom, and the other Slater determinant (= Slater-2 ) contains A electron with up-spin and B electron with down spin inside He atom.
↑ The up-down spins of A and B electrons inside He atom were swapped between two determinants that can generate artificial singlet between A and B electrons ( +∫φA ↓(1) φB ↓(1) = positive attractive exchange energy between two wavefunctions A and B with the same down-down spin which integral does not vanish, ← integral of spin up-down vanishes ).
The total wavefunction of Slater-1 determinant minus Slater-2 determinant can generate the singlet (= positive ) attractive exchange energy between A and B electrons with up-down spins (- correctly, changing the original two-up-down spins inside He into two up-up or down-down spins between two Slater determinants ) inside He atom (= But A and B belong to the same He 1s wavefunction, so actually No exchange energy occurs between the singlet A and B up-down spins, so meaningless ).
↑ In this case, only Pauli repulsive (= negative ) exchange energies occur between A electron of He and C electron (= φC ) of H, and between B electron of He and C electron of H, which causes unnecessary Pauli repulsion between He and H atoms due to each antisymmetric Slater determinant (= which case corresponds to this-middle-Symmetry of three-electron wave functions ).
If we try to artificially generate the molecular attractive ( positive ) exchange energy also between A electron with down spin of He and C electron with up spin of H atom, and swap the down-spin of A electron and the up-spin of C electron, the He atom ends up containing A electron with up spin and B electron with the same up spin, and this He atom with two same up-up spins vanishes by Pauli antisymmetric wavefunction inside one Slater determinant ( this figure-lower ).
So it is impossible to make artificial singlet attractive exchange energy between A electron of He and C electron of H atom even by using multiple Slater determinants.
As a result, we can prove quantum mechanical antisymmetric wavefunctions or Slater determinants are false, unable to describe molecular bond attractive exchange energy between different atoms.
See DFT is useless.
(Fig.7) Four Helium atoms (= A,B,C,D ) are arranged tetrahedrally. ← Each unphysical molecular orbital ( in four He1, He2, He3, He4 molecular orbitals ) contains two electrons up and down spins: each electron must be unrealistically spreading all over four Helium atoms.

Here we show one typical example where the unphysical quantum mechanical molecular orbitals can neither remove unnecessary Pauli repulsion nor generate the necessary van der Waals attraction between four helium atoms (nor generate covalent sigma bond's energy in molecules with multiple atoms, this p.1-left-2nd-paragraph ), hence, quantum mechanics is inconsistent with the experimental results, and wrong.
In the upper figure, four helium atoms (= each of He-A, He-B, He-C, He-D atoms contains each helium 1s atomic orbital wavefunction φA, φB, φC, φD, respectively ) are arranged regular tetrahedrally (= distances between any two helium atoms are supposed to be equal, meaining all the overlap or exchange integrals (= S ) between any two of these helium atoms are also equal ).
Each atomic orbital contains one electron, which condition is called "normalization" expressed as the integral of two same atomic wavefunctions being 1 like
∫φAφA = ∫φBφB = ∫φCφC = ∫φDφD = 1
And the overlap (= or exchange ) integral between any two different helium atomic orbital wavefunctions becomes S due to the same interatomic distance between any two electrons ( this-(7) ) like
∫φAφB = ∫φAφC = ∫φAφD = ∫φBφC = ∫φBφD = ∫φCφD = S.
Four helium atoms contain the total eight electrons = four electrons with up spins and four electrons with down spins.
Due to the fatal flaws of quantum mechanical Pauli antisymmetric wavefunctions, any molecular bond attractive exchange energies between two different up and down spins become zero, and only unnecessary Pauli repulsive exchange energies between two electrons with the same up-up or down-down spins remain.
↑ To avoid this quantum catastrophe, quantum mechanical ad-hoc molecular orbital theory demands that any overlap or exchange integrals between any two different molecular orbitals must be zero, which artificial condition is called "orthogonal" or "orthonormal (= orthogonal + normalization, this p.14, this p.6 )."
Each molecular orbital can contain the maximum two electrons with up and down spins due to Pauli exclusion principle, so four different molecular orbitals (= He1, He2, He3, He4 ) are necessary to describe interactions between four helium atoms containing eight electrons (= two electrons with up and down spins × four molecular orbitals ).
Quantum mechanical molecular orbital is unrealistic, because each electron must always spread all over four helium atoms, and each electron's unphysical molecular orbital (= MO ) must be expressed as the linear combination of multiple (= four helium ) atomic orbitals (= AO, this middle-methane-case ).
Each molecular orbital contains one electrons, so the ( Coulomb ) integral of the same two molecular orbital (= total probability density of each electron ) must be 1, like
∫He1He1 = ∫He2He2 = ∫He3He3 = ∫He4He4 = 1
which normalization condition gives different normalization coefficients to different molecular orbitals.
As shown in the upper figure, we can find four (unphysical) molecular orbitals which are orthogonal to each other, which means the overlap integrals of any two different molecular orbitals must be zero like
∫He1He2 = ∫He1He3 = ∫He1He4 = ∫He2He3 = ∫He2He4 =
∫He3He4 = 0.
↑ But even if this artificial orthogonal molecular orbitals are created, quantum mechanical molecules cannot avoid unnecessary Pauli repulsive exchange energies between the same spins (= between four electrons with the same up spin of four helium atoms, and between four electrons with the same down spin of four helium atoms ), after all, so quantum mechanics is false.
Due to the orthogonal molecular orbitals (= any overlap or exchange integrals between any two different molecular orbitals are zero ), only ordinary Coulomb integrals (= integral of two same molecular orbitals ) remain, when we integrate the chosen wavefunctions with Schrodinger energy equation (= H ) to obtain (fake) total energy.
↑ But each molecular orbital is the linear combination of multiple different atomic orbitals, so the ordinary Coulomb integrals of the same molecular orbitals or the square (= probability ) of each molecular orbital automatically contain the exchange energy integrals.
The nomalization coefficients of Pauli repulsive antibond molecular orbital including the minus-phase atomic orbital tend to be bigger than the attractive molecular bond orbital including only the positive-phase (spatial) atomic orbitals.
So after all, the sum of the ordinary Coulomb integrals of four molecular orbitals gives the Pauli repulsive exchange energy integrals (= negative overlap or exchange integral = -S ) between the above four helium atoms.
It means the quantum mechnaical molecular orbitals can Not avoid unnecessary Pauli repulsive exchange energies, and cannot express the actual van der Waals attraction between helium atoms without adding artificial pseudo-potential energies. ← Quantum mechanics is proven wrong.

Feel free to link to this site.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
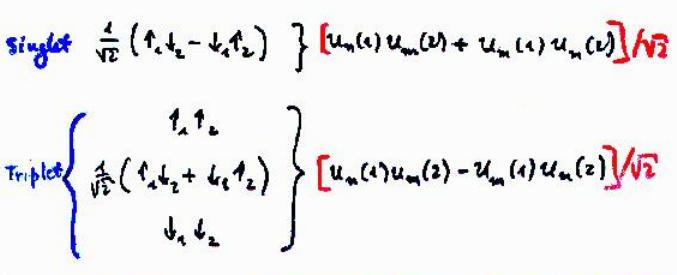{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}