
Home page
Quantum mechanics is useless
(Fig.1) Quantum mechanics just artificially adjusting free parameters cannot predict any atomic energies.
We have been brainwashed into falsely thinking quantum mechanics with parallel worlds is useful for modern technology by all the media, academia and corporations for long years.
Actually, quantum mechanics cannot predict anything, much less contribute to any technologies ↓.
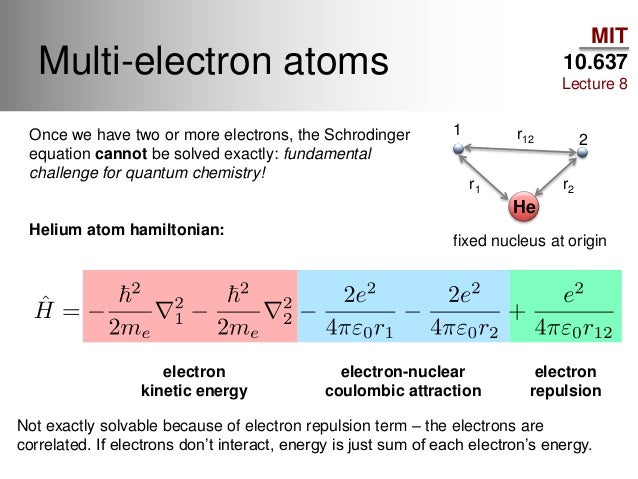
Quantum mechanics relies on solving Schrödinger equations where the conserved constant total energy E is the sum of electrons' kinetic energy expressed as differential operators acting on atomic wavefunctions and Coulomb potential energy among electrons and nuclei.
But quantum mechanical Schrödinger equation for only one-electron hydrogen atom can be solved, which solution agrees with successful Bohr atomic model ( this-p.12, this-4.133 ).
Quantum mechanical Schrödinger equations for multi-electron atoms such as helium and hydrogen molecule ion (= H+ ) are unsolvable, unable to have true wavefunctions ( this-p.21, this-p.4-1st-paragraph ), so quantum mechanics cannot predict energies of any multi-electron atoms or molecules.
↑ The fact that No wavefunctions can become true solutions for multi-electron atomic Schrodinger equations conserving the constant total ( ground-state ) energy E (in any different electrons' positions ) shows quantum mechanical Schrödinger equations and wavefunctions proved to be false ( this p.2-upper, this p.7 ).
For unsolvable Schrödinger equations that cannot predict anything, quantum mechanics has to choose fake trial wavefunctions (= basis sets ) out of infinite free forms and infinite free parameters ( this-p.9-last, this-p.2-right-upper ) to find ones giving the lowest energy, which takes infinite time, impractical, cannot predict anything in its approximate variational methods ( this-p.3(or p.1 ), this-p.14,p.25 ).
This-1st~5th-paragraphs say -- Freely choosing fake solutions
"The variational method is one way of finding approximations to the lowest energy eigenstate or ground state. The method consists of choosing a "trial wavefunction" depending on one or more parameters, and finding the values of these parameters for which the expectation value of the energy is the lowest possible"
"The trial wavefunction is selected to be a function of one or more parameters that resemble quantum numbers in the true wavefunctions, but are NOT obtained from solving the Schrödinger Equation (i.e., there is 100% freedom in selecting the form of trail wavefunction). There is no limit to the number of parameters the trial wavefunction may have."
It is impossible to find fake trial wavefunctions giving the lowest energy out of infinite free choices for unsolvable Schrodinger equations, so quantum mechanical approximate variational methods also cannot predict any atomic energies ( this or this-p.7-caveat, this-(9.80), this or this-p.4-1st-paragraph ).
This or this-p.1-last says -- No quantum mechanical prediction
"The theorem (= quantum mechanical approximate variational methods ) does not tell us how far above the ground state energy the left hand side of the
inequality (1) is, so it would not seem to be of much help in estimating the ground state energy."
This-p.3-last says -- Useless quantum mechanics
"Problem (of quantum variational methods ): you do NOT know how
close your result is compared to the exact result. You
only know you are above"
↑ So the ab-initio or first-principle quantum mechanics is fake, unable to predict anything ( this or this-p.6-2nd-paragraph ) and are proven wrong, contrary to a lot of hyped news.
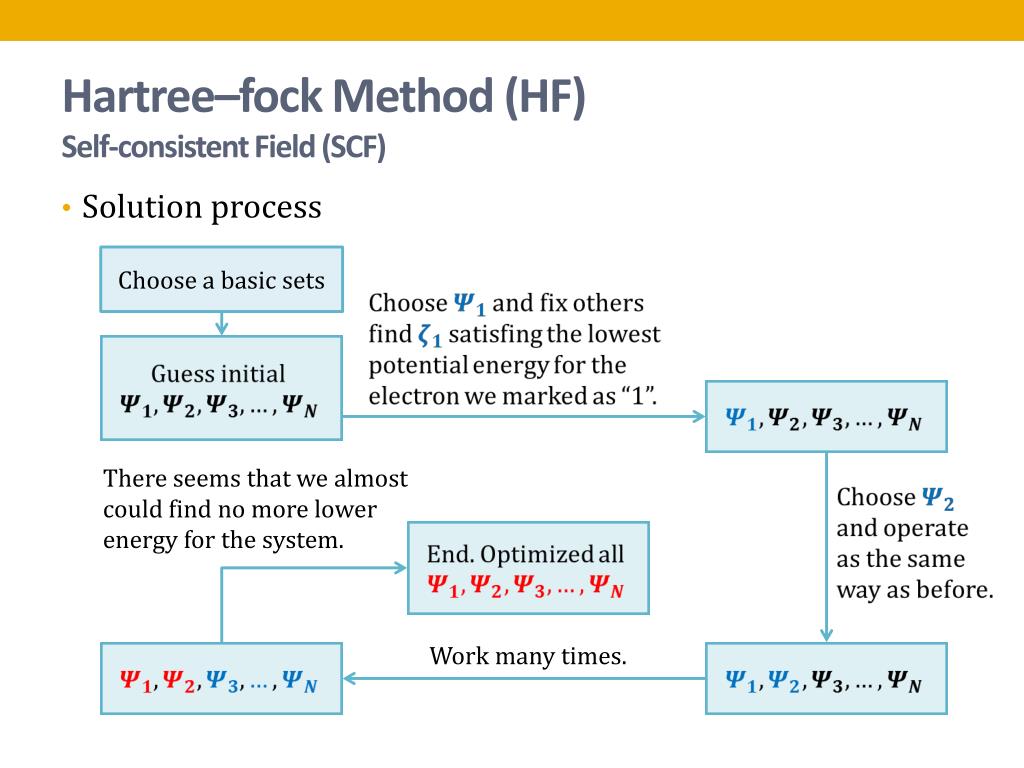
In this impractical quantum mechanical approximate variational methods such as Hartree-Fock ( this-p.14-last ) and configuration interaction (= CI, this-p.2-introduction ), finding coefficient free parameters of the artificially-chosen fake trial wavefunctions giving the lowest energy by too time-consuming self-consistent method (= SCF ) is misleadingly called " solving ( this-p.6-p.7, this or this-1.4 )", which is Not true solving the unsolvable Schrödinger equations ( this-p.14 ).
For unsolvable Schrödinger equations, quantum mechanics relies on impractical approximate methods such as perturbation and variational methods.
This quantum approximate perturbation methods try to express solutions for unsolvable Schrodinger equations by infinite terms or series by choosing fake wavefunctions or basis sets, which are impractical, too time-consuming, cannot predict anything.
Of course, calculating infinite terms is impossible. So physicists often choose only a few terms of fake trial wavefunctions that can give wrong energies lower than true atomic ground-state energy ( this-p.27, this-p.8-6th-sentence ).
And No guarantee that the chosen trial wavefunctions of infinite terms can converge to some values ( this-p.15-1st-paragraph, this-p.27, this-p.1-intro-1st-paragraph ).
So this impractical quantum mechanical approximate perturbation is less accurate and less popular than variational methods ( this or this-p.22-last-paragraph, this-p.6-remark, this-p.5-last, this-last ).
(Fig.2) Quantum mechanics has to artificially choose free parameters to adjust calculated (fake) atomic energies, which cannot predict any true atomic energies.

For unsolvable Schrödinger equations that cannot predict anything, quantum mechanics can only choose fake trial wavefunctions out of infinite free choices and infinite free parameters to find ones giving the lowest energy in its approximate variational methods ( this-p.22-5.1 ).
This quantum mechanical variational methods says any chosen (fake) trial wavefunctions give energies higher than true atomic ground-state (= lowest ) energy ( this-p.15-middle, this-p.3-last,p.5-lower ), which is actually false, disproving quantum mechanics.
In fact, in these quantum mechanical ( variational ) methods for the unsolvable useless Schrodinger equations, physicists just deliberately manipulate free parameters (= using only a few exponent parameters k, ζ, of e-kr, e-ζ r..) of artificially-chosen fake wavefunctions to seemingly obtain (fake) total energy close to experimental value ( this p.6-p.7 roughly fixed exponent parameters ζ to 6.50 or 40.5 without rigorously optimizing them ), which is Not prediction of quantum mechanics.
This p.6-IV says the exponents are considered as adjustable parameters.
↑ Getting values close to experimental atomic energy by using only a few (exponent) free parameters fixed to some values means quantum mechanics and its variational methods are wrong, because if they more rigorously optimize more free parameters, these variational methods (= supposed give only higher energies than true atomic energies ) can give wrong energies lower than the true atomic ground-state (= lowest ) energies.
Only in a two-electron Helium (or lithium ) among the unsolvable multi-electron atomic Schrodinger equations, physicists artificially choose Hylleraas-type (fake) trial wavefunctions including variables of interelectronic distance (= r12 = distance between two electrons-1 and 2, this-p.2 ), which can give (fake) total energies lower than other trial wavefunctions in variational methods.
The problem is that the calculation (= integral ) of this chosen Hylleraas trial wavefunctions is impractical, too time-consuming to apply to atoms with more than four electrons.
↑ It means quantum mechanics can Not obtain energies matching experimental energies of atoms with more than three atoms, even in its illegitimate approximate (= variational ) methods ( this or this-p.18(or p.16), this-p.1-right-upper ).
(Fig.2') Quantum mechanics just choosing free parameters cannot predict atomic energies.

Here we show that the current quantum mechanical (variational) methods relying on freely-adjustable parameters (= instead of finding the parameters giving the lowest possible energy ) is useless, wrong, can Not predict (Helium) atomic energies.
For artificially getting helium's ground-state energry (= -2.903724.. Hartree without reduced mass, this-p.6 ), physicists used only a few exponent free parameters roughly or randomly fixed to some values, which means quantum mechanics and its variational methods are wrong, because if they more rigorously optimize those parameters, these can give wrong energies lower than actual helium ground-state (= lowest ) energy. ↓
In this paper about the current best quantum variational method for seemingly getting actual helium ground-state energy ↓
p.1-(1)(2), p.3-Table.I says "where αi, βi and γi are complex (free) parameters (= exponent parameters of the artificially-chosen helium Hylleraas wavefunctions ) generated in a quasi-random manner (= instead of optimizing those parameters variationally to give lower energies, they fixed these exponent parameters to some desired round numbers with No quantum prediction, this-p.3-Table I-A, B, G.. ).
↑ Just adjusting parameters by quasi-random manner giving the helium ground state energy means they can give wrong energies lower than true atomic energies, if they really rigorously try to find exponents parameters giving as low energies as possible out of infinite parameter choices, which means quantum mechanics and its variational methods are wrong."
This paper-p.12-2nd-paragraph says -- Artificially-fitting parameters
"Also, I know of no published theoretical attempts to explain the excellent convergence found by Korobov with his highly nonlinear fitting of the
trial wavefunction..
Korobov’s approach (= upper-quasi-random manner ) may be likened to the work of fitting the Hydrogen
radial wavefunction with a set of gaussians, using floating exponents.
This sounds plausible, but at present it is just more handwaving about flexibility."
This or this-paper (= one of today's best quantum variational methods for seemingly getting helium ground-state energy ) ↓
p.4-left-last-paragraph also says -- Free exponent parameters
"The orbital exponent may be treated as a nonlinear (= exponent ) variational parameter,
though this is Not strictly necessary (= they don't try to find the lowest energy by using exponents as strictly-variationally-optimized parameters )"
p.5-(24), p.9-Table.V, p.10-E -- Wrong variational method
They intentionally fixed one non-linear parameter β = 1 and another exponent parameter α = 2.20 (= if they more strictly vary these exponent parameters, they could find fake energies lower than true ground state energies, which disproves quantum mechanical variational method that must always give energies higher than true atomic energies ).
Schwartz intentionally fixed an exponent parameter k to 3.5 (= k is Not used as a variational parameter finding the lowest energy, this-introduction-(3), p.1147-Results says the parameter k was fixed at the artificially-chosen value of 3.5 ) to illegitimately obtain Helium ground-state energy by quantum variational method.
Also in the recent calculation, he roughly fixed the exponent (= scaling ) parameter k at 2.0 ( this p.2-(0.3) ) to seemingly get exact helium ground-state energy, which means if they more rigorously optimized more free (exponent) parameters, they could get wrong energy lower than actual helium ground-state (= lowest ) energy, disproving quantum mechanics.
Another research also intentionally fixed exponent parameters (= η, ζ ) of helium Hylleraas wavefunctions at (artificially-chosen values of) 6.00 or 6.50 ( this-p.6-lower~p.7-Table.1 ) to deliberately prevent their calculated energies from being lower than true helium ground-state (= lowest ) energy.
Drake also treated the exponent nonlinear parameters as freely optimized scale factors ( this p.4-Table.IV ).
↑ It means quantum mechanical variational methods can get wrong energy values lower than true ground state energies (= in atoms such as helium ) if physicists use various forms of wavefunctions (= including interelectronic distance like Hylleraas ) and really try to find parameters giving the lowest possible energies.
↑ This fact of chosen wavefunctions giving wrong energies lower than true atomic energies disproves quantum mechanics or its variational method choosing (fake) trial wavefunctions that must always give energies higher than the true atomic lowest ground-state energies.
They intentionally fixed those (exponent) parameters at some round numbers (= this-p.2-(0.3) roughly fixed one exponent scale parameter at k = 2.0, this-p.2-(6).p.3-(7)(8) also used roughly-fixed exponent parameters of γ = 2.0, 1.2.. ) to prevent the calculated values from being lower than actual helium ground-state (= lowest ) energy.
↑ As a result, quantum mechanics and its variational methods are proven wrong, giving wrong energies lower than true atomic ground-state ( = lowest ) energies if they really rigorously optimize exponent free parameters.
This or this-p.1-introduction-4th-paragraph (and p.5-Table.1 ) says "However, according to recent experimental result (Bergeson et al., 1998), the results from the techniques reported above overestimated the helium ground-state energy" ← Quantum mechanical variational methods turned out to be Not variational, so false.
↑ This-p.2-(0.3) shows physicists intentionally fixed exponent (= scaling ) parameter k = 2.0 to get desired helium ground-state energy values without rigorously varying k variationally, which disporves quantum mechanics and its variational methods.
↑ In fact, not only exponent parameters but also linear variaitional parameters = coefficient C of trial wavefunctions were artificially chosen to get the desired helium ground state energy with No quantum mechanical prediction. ↓
This-p.245-II ~ p.246 (or p.245 )-(5), p.243-(2) also tried to artificially choose coefficient variational parameters C based on artificially-chosen F function converging to some desired helium ground-state energy, which is Not quantum mechanical prediction nor variational methods.
Not only in helium's ground-state energy, but also in helium's excited energy, quantum mechanical variational methods can only artificially choose exponent free parameters in a pseudo-random way instead of rigorously optimizing those parameters, which is Not prediction.
This-p.4(or p.3)-last-paragraph says -- Fake variational method
"a variational approach based
on exponential expansion with the parameters of exponents being chosen in a pseudorandom way"
Also in hydrogen molecule (= H2 ), quantum mechanical variational methods can only artificially fix a few exponent parameters to some chosen values instead of rigorously optimizing those parameters.
This-p.7 shows the H2 molecule's trial wavefunctions' exponent parameter δ was artificially fixed to 0.75.
This-p.3-p.5 shows they artificially fixed exponent parameters α of H2 molecular trial wavefunctions to two chosen values of 1.32075 and 6.32075.
↑ If they more rigorously optimize more exponent parameters, they could get wrong energy lower than true H2 molecular ground-state lowest energy.
So quantum mechanics and its variational method supposed to give only upper bound to true energy are proven wrong.
Also in lithium case, quantum mechanics just chooses artificially-fixed exponent parameters like helium ( this-p.2-right-last-paragraph ), which disproves quantum mechanics and its variational method.
Quantum mechanical Pauli antisymmetric wavefunction is wrong, cannot be applied to more than two electrons like lithium.
The lithium wavefunction (= this-(4) ) contains only one symmetric relation between electrons 1 and 2 (= attractive exchange energy ), while other relations between electrons 2 and 3, and electrons 1 and 3 are antisymmetric (= Pauli repulsive exchange energy ).
↑ This is wrong, because if the relation between two electrons with spin up (= α ) and down (= β ) is symmetric in three electrons with up, up, down spins, not only relation between electrons 1 (= up spin ) and 2 (= down spin ), but also relation between 2 (= down spin ) and 3 (= up spin ) must be symmetric.
This-lithium energy calculation ↓
p.4-3rd~4th-paragraphs say -- Only 5 parameters used
"We noticed that the use of only one set of wi's (= exponent parameter ) does not lead to accurate results, therefore,
following Yan and Drake, we divide the whole basis set into 5 sectors, each one with its own
set of wi's"
p.4-last-paragraph says -- Wrong prediction
"The result for the nonrelativistic energy is significantly below the previous estimate
and indicates that extrapolation to infinite basis length does not always give the right result"
↑ So this research used only 5 exponents variational parameters (= coefficients parameters were used as free parameters ) to get lithium energy, which means if they used more exponent parameters, they could get wrong lithium energy lower than the true ground-state lowest energy, which disproves quantum mechanics.
↑ Furthermore, this calculation of lithium energy does not minimize energy with respect to coefficient parameters (= C ) ( this-p.3-p.4 ) in a too time-consuming self-consistent way (= often fail to converge ) with the exponent parameters (= it is impossible to minimize total energy with respect to both coefficient and exponent parameters at the same time ), which means they use the coefficient parameters as free parameters, which is Not quantum mechanical prediction nor variational methods.
This-p.5-Table 1.-lower says
"K-shell orbital exponents are K = 3.5, Kp = 4.1 ; L-shell orbital exponents are L = 0.78237, Lp = 1.0375"
↑ So quantum mechanics variational methods just artificially fix exponent parameters to some chosen numbers with No ability to predict Beryllium's ground-state or excited energy.
For unsolvable Schrodinger equation that cannot predict anything, quantum mechanics has to choose fake trial wavefunctions out of infinite candidates which must include variables of interelectronic distance (= r12 ) as shown in Hylleraas wavefunctions to find ones giving the lowest total energy.
But using these Hylleraas wavefunctions with variables of interelectronic distance is too time-consuming and impractical in atoms with more than 4 electrons ( this-p.3-introduction ).
So quantum mechanics even officielly cannot get true energies in atoms with more than 4 electrons, hence, they are useless for any technology such as transistors, MRI, sensors, all of which involve atoms and molecules with more than 4 electrons.
(Fig.3) Quantum mechanical unsolvable Schrodinger equations have to artificially choose fake unphysical trial wavefunctions in the impractical CI.
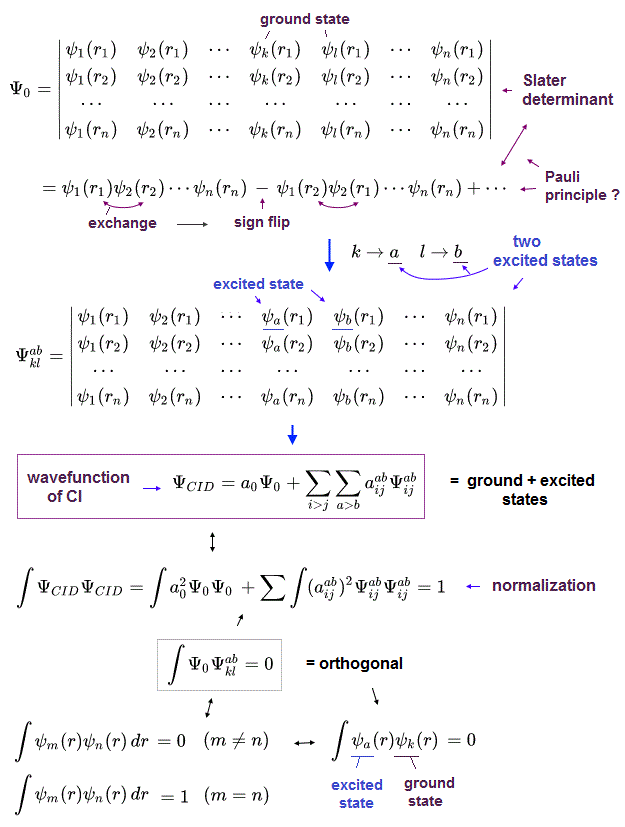
Quantum mechanical ( artificially-chosen ) wavefunctions (for unsolvable useless Schrodinger equation ) obeying the ad-hoc Pauli principle must be expressed as (unphysical) antisymmetric wavefunctions or Slater determinants, whose Schrodinger energy approximate calculation is called Hartree-Fock (= HF ) equations ( this p.6 ).
These quantum mechanical approximate Hartree-Fock (= HF ) or molecular orbital methods are known to be unable to give the total energies close to experimental values even in their chosen trial wavefunctions. ← the difference between this Hartree-Fock equation and true energy is called correlation energy.
The current most-widely used quantum mechanical approximate methods to seemingly get this correlation energy beyond Hartree-Fock is one-pseudo-electron density functional theory (= DFT ) that just artificially chooses fictitious exchange-correlation (= XC ) energy functional and pseudo-potential ( this or this-p.3-9 ), so DFT is fake ab-initio unable to predict any values.
The unsolvable useless quantum mechanical Schrödinger equations, which cannot predict anything, have to rely on the impractical time-consuming approximate variational methods called configuration interaction (= CI or full-CI or FCI, this-p.5-last, this-p.2-introduction, this-p.6-lower ), which cannot predict anything ( this or this-p.4-1st-paragraph ).
In this impractical (full) CI, the artificially-chosen trial (fake) wavefunctions (= ΨCID ) are expressed as the linear combination of more than one ( unphysical ) antisymmetric Slater determinants ( this-p.3-4, this-p.21 ).
This impractical quantum mechanical approximate CI chooses fake trial wavefunctions containing fictional excited atomic wavefunctions ( this-p.8, this-p.8 ) that are impractical, too time-consuming, cannot predict anything.
↑ The main mechanism for this CI to (illegitimately) lower total energy is the (unreal) exchange energies between (artificially-excited) electrons ( this-p.30-37, this-6.5.4~ ).
Because the artificially-chosen excited and ground-state trial wavefunctions are often orthonormal to each other (= integral of these wavefunctions are artificially made to be zero ), which cannot make the unphysical exchange energy between two (fictitious) electrons zero ( this or this-p.1,7, this-p.3 ).
↑ Unphysical exchange energy between two electrons caused by the chosen fake trial wavefunctions' Slater determinants can illegitimately lower the total energy in CI
↑ Even this impractically time-consuming full CI cannot give (fake) total energies matching experimental values, because multi-electron atoms with more than three electrons cannot use Hylleraas wavefunctions that can give wrong energies lower than the true atomic energies, which disproves quantum mechanics.
(Fig.4) Coupled cluster (= CC ), which is Not variational, can give wrong energies lower than true atomic lowest ground state energy, so CC is Not a right theory.
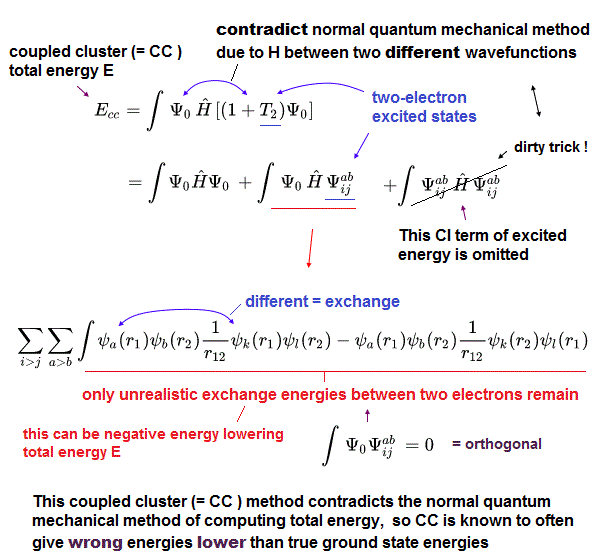
Instead of impractical time-consuming configuration interaction (= CI, this-p.6-left, this-p.2-right-A ), one of the impractical time-consuming approximations called coupled cluster (= CC or CCSD ) theory is often used ( this or this-p.7-right ).
For unsolvable Schrodinger equation that cannot predict anything, this coupled cluster (= CC ) theory ( this-p.2-introduction ) can choose fake trial antisymmetric wavefunctions (= basis sets ) illegitimately combining Hartree-Fock (or CI ) Slater determinants (= Ψ ) and their virtual excited-energy orbitals ( this-p.2 ) which also cannot predict anything.
This or this-lower-Computational cost says -- CC is too time-consuming
"it would take 120 years to perform the same calculation on a cluster of 20 water molecules."
These Coupled cluster (= CC ) fake trial wavefunctions can give wrong energies lower than the true ground-state lowest energies, which means CC is intrinsically a false theory (= CC is Not variational method, though the orthodox quantum variational method is also wrong ).
This or this-(6.30) says -- CC = Not variational
"The result is a set of equations which yield an energy that is not necessarily variational (i.e., may Not be above the true energy)" ← Coupled cluster (= CC ) is false.
This p.1-left-1st-paragraph says -- CC is wrong
"it is worth noting that
CCSD (= coupled-cluster with singles and doubles ) is Not variational, and it is also Not guaranteed
to give real-valued energies"
This or this p.16-top says -- CC wrong energy
"Consequently, the total CC energy is Not necessarily above the
exact ground-state total energy, just as in perturbation theory"
In CC, total energy (= E ) is given in a wrong way putting Hamiltonian energy equation (= H ) between two different wavefunctions (= artificially-chosen, cannot predict anything ) like E = ∫Ψ0HΨab (← Not a normal ∫Ψ0HΨ0 or ∫ΨabHΨab, this p.18-(3.15), this-p.15-(82), this or this p.27-2.29, p.29-2.40, this-6.28, this-p.8-(17) ), which is the trick of CC lowering the total energy illegitimately faster than CI.
The (fake) total energy of the illegitimate CC can be lowered by the unphysically-negative exchange energy between two electrons of ground state and excited state (= ∫ψaψb1/r12 ψkψl, this p.8, this-p.7-lower, this-lower, this-(4) ) like CI.
↑ This means CC deliberately omits the annoying positive excited energy part (= ∫ΨabHΨab ) to illegitimately lower the total energy than CI.
So CC is known to be able to get total energy lower than true ground state energy (= CC is Not variational ), which fact invalidates CC method.
There are many cases where coupled cluster (= CC, CCSD, CCSDT ) disagreed with experimental results.
This p.5-3rd-paragraph says -- CC is wrong
"The CCSD model systematically overestimates all core excitation energies".
This p.36-3rd-paragraph says -- CC fails
"CC fails for systems
with strong static correlation"
This-abstract-last says -- CC wrong energy
"CCSD(T) overestimates the formation energy"
This p.6-left-2nd-paragraph says -- CC wrong energy
"CCSDT we observe partially strong overestimation of the
total energy ( this-p.5-left )"
(Fig.5) MP2 is not variational, because it can give wrong energies lower than true atomic energy illegitimately.
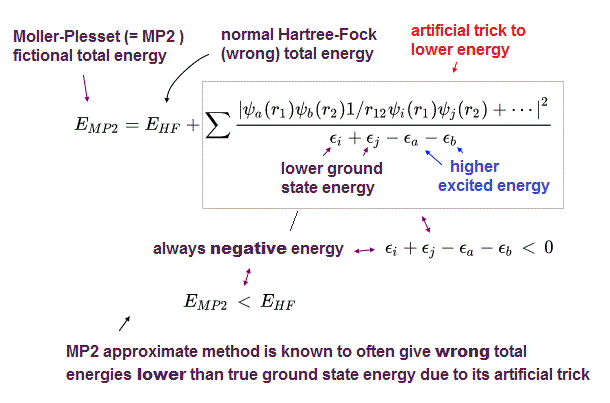
Instead of extremely-time-consuming configuration interaction (= CI ) and coupled cluster (= CC ), one of the illegitimate quantum mechanical approximations called Møller–Plesset perturbation theory (= MP) is often used to give the total energy lower than Hartree-Fock (= HF ) energy using deceitful tricks.
As shown in above figure, MP2 total energy (= EMP2 ) is always lower than Hartree-Fock energy (= EHF ) due to the artificially-introduced second term that is always negative.
The problem is this MP or MP2, which perturbation method (= PT ) is Not variational theory, which often gives wrong energies lower than true ground state energies (= overestimation of energies, this abstract-last, this p.7-2nd-paragraph ), which means Møller–Plesset perturbation theory is an unreal theory.
This p.12-last says -- MP2 wrong
"the MPn methodology is Not variational – it is possible that the
MP2 estimate for the correlation energy will be too large instead of too small."
This p.10-last says -- MP2 = Not variational
"MP theory uses a truncated Hamiltonian, and is therefore Not
variational. That means that one cannot use the calculated energy as a measure
of the quality of the calculation."
This or this-introduction-2nd-paragraph says -- MP2 fails
"Despite this popularity, the limitations of MP2-like methods are also well-known. In particular, they fail spectacularly for strongly correlated or metallic systems. Furthermore, a strong overestimation of dispersion interactions is observed for large polarizable systems"
This 1st-paragraph says -- MP2 wrong
"it suffers from the usual MP2 overestimation of dispersion."
This or this-p.2-2nd-last-paragraph says -- MP2 wrong
"Thus, the value of E(2), the first perturbation to the Hartree-Fock energy, will always be negative. Lowering the energy is what the exact correction should
do, although the Møller-Plesset perturbation theory correction is capable of overcorrecting it, since it is not variational"
↑ As shown here, Møller–Plesset perturbation theory (= MP2 ) is a defective illegitimate theory.
As a result, all the quantum mechanical methods are wrong, illegitimate and useless theories which must be replaced by the realistic simple useful atomic model.

Feel free to link to this site.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}