トップページ (2電子原子も含む正確な新ボーア模型)
タンパク質構造のページに戻る。
電子スピンは幻想である。
ブラックホールは幻想である。
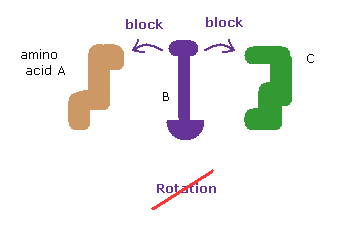
(Fig.1) 回転が両方向にブロックされている。
タンパク質の構造は 二面角 (= ねじれ角 ) で決まる。
各回転部位で、アミノ酸は ある範囲に自由に回転できる (= 回転異性体 )。
このウェブサイトでは 2つの 離れたアミノ酸の衝突が これらの回転をブロックして その結果 タンパク質全体の構造が決まることを示す。
つまり タンパク質は 回転する固体のように振舞うと言える。
ここでは タンパク質構造を調べたり 操作するのに 次の サンプル JAVA プログラムを使用する。
サンプル JAVA プログラム ( PDB タンパク質 - 回転 )。
最初に 次のミオグロビンを扱う。
Oxymyoglobin text ( 1MBO.txt ).
また このプログラムは 各アミノ酸側鎖を変化させることができる ( 例えば、 ALA → PHE など )。
そのため 次のテキストファイルを 19 種類のアミノ酸のテンプレートとして使用する。
(Fig.1) セーブフォルダー位置の変更。

このテキストを "C:\sdff" フォルダーの位置に "1MBO.txt" のファイル名でセーブする。
( もしくは PDB ウェブサイトから 直接セーブすることもできる。 )
同じように "tout.txt" のテキストも フォルダーにセーブする。
( 違うフォルダーにセーブした場合は、プログラム内の Fig.1 部分を変更するように。 )
その後、上の サンプル JAVA プログラムを "amic.java" としてセーブしてコンパイルする。
"詳細は、-Xlint: unchecked オプションを指定して再コンパイルしてください。" などの注意は無視してそのまま実行できる。
(Fig.2) タンパク質画面。
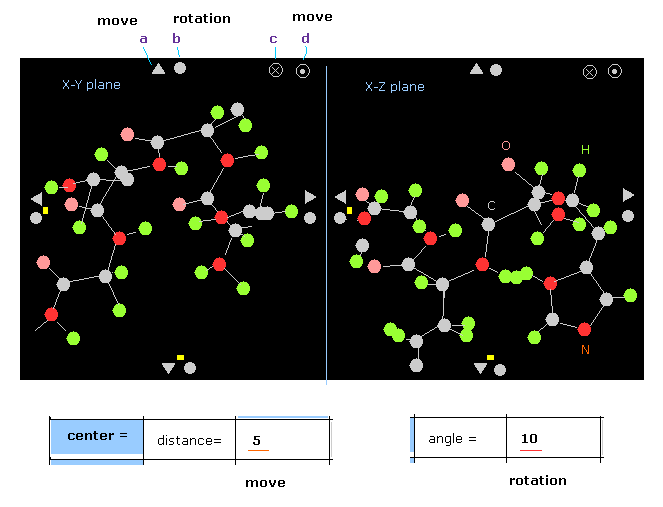
基本的な操作方法は このページと ほぼ同じである。
ある 三角形 (= a ) をクリックすると、 すべてのタンパク質は その方向へ移動する。
ワンクリックにつき 移動する距離は "distance =" の横の数値に依存する。
Fig.2 のケースでは、上の三角形を クリックすると、タンパク質は +y 方向へ 5.0 Å 移動する。
( この値が 10 より大きいと、 移動距離は 2.0 Å になる。)
ある 円部分 (= b ) をクリックすると、タンパク質全体は その方向へ回転する。
ワンクリックにつき 回転する角度は "angle =" 横の数値に依存する。
Fig.2 のケースでは、 回転角度は 10°となる。
x-y 平面上で "x " マーク (= c ) をクリックすると、すべての分子は -z 方向へ移動する。
( x-y 平面上で "点と丸" のマーク (=d ) をクリックすると、 z 方向へ移動する。 )
(Fig.3) コマンドプロンプト画面。
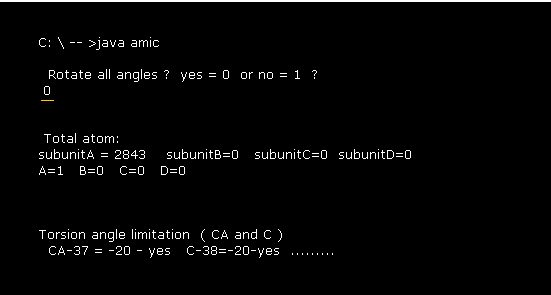
プログラム実行後、Fig.3 のメッセージが コマンドプロンプト画面上に表示される。
ここで "0" を入力すると、 このプログラムは CA と C の すべての 二面角を計算する。これに関しては 後で説明する。
この 1MBO 蛋白の 付加した水素原子を含んだ原子総数は "2843" である。
1MBO は 1 サブユニット (= A のみ ) で構成されている。
(Fig.4) テキスト画面。
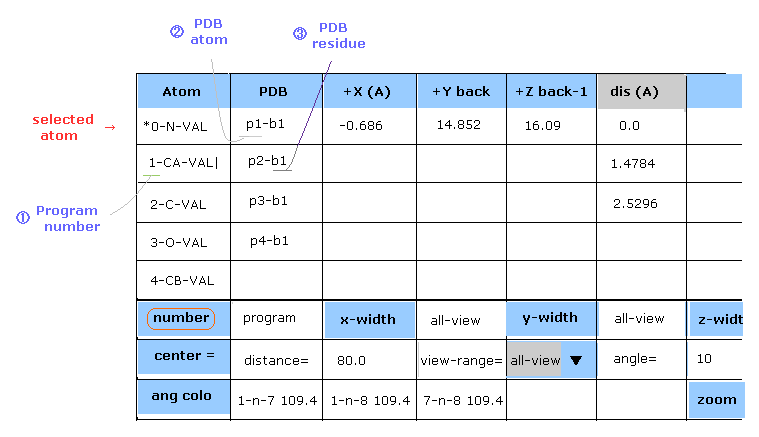
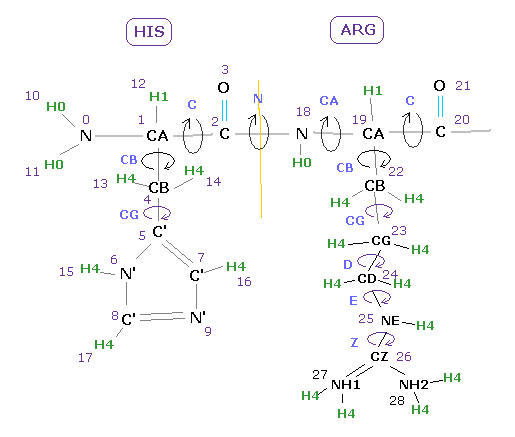
このプログラムでは 3つの原子番号が使われている。
1つめは ( プログラム ) 原子番号で これは 水素原子を含むすべての原子に与えられる。
2番目が PDB 原子番号で、3つ目が PDB 残基 (= アミノ酸 ) 番号 である。
( 水素原子には PDB 原子番号がついていない。)
(Fig.5) PDB データファイル例。

"number" ボタンをクリックすると、画面上に表示されている 原子番号の種類を変更できる。
( 個人的には PDB の残基番号が見やすいと思う。 )
"B" の文字が B サブユニットの原子についている。
(Fig.6) 二面角 ( ねじれ角 )。
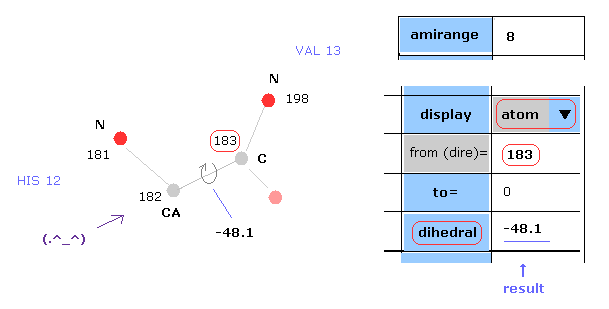
二面角 は それが決まれば タンパク質全体の構造が決まるため 非常に重要な概念である。
二面角は Fig.6 において 平面 A (= 181-182-183 ) と 平面 B ( 182-183-198 ) 間の角度を表している。
スクロールバー内から "atom" を選択して、 "183" の数値を "from (dire)=" の横のテキストボックスに入力して "dihedral" ボタンをクリックすると、ここの二面角 ( 182 と 183 間の ) を得ることができる。
"atom" を選択しているときは プログラム原子番号で指定し、 "PDBat" を選択しているときは PDB 原子番号を指定する。
(Fig.7) "residue" (残基)+ "C" の選択。
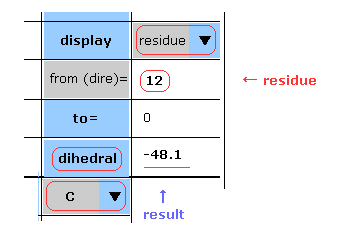
各スクロールバー内から ( N, CA, C, CB, CG, ... ) のどれかと "residue" を選択しても二面角を得ることができる。
この場合、 "C" を選択して PDB の残基番号 (= HIS-12 ) をテキストボックス内に入力して "dihedral" ボタンをクリックする。
( 基本的に ペプチド結合は 二重結合性のため、 "N" の場合は、この角度は -180°近くになる。)
水素原子や酸素原子は 二面角に何も関係がないことを注意しておく。
(Fig.8) 二面角と原子の選択。.
例えば、Fig.8 で CA 直前の 18-19 間の二面角が知りたいときは 原子番号 "19" をテキストボックス内に入力して "dihedral" ボタンをクリックする。
基本的に CA や C の炭素の直前の角度は ラマチャンドランプロットとして知られ、αへリックスやβシートの構造にとって 非常に重要である。
"4" (=CB ) を入力すると、Fig.8 において 平面 0-1-4 と 平面 1-4-5 間の角度を知ることができる。
"26" (=CZ ) を入力すると、平面 24-25-26 と 平面 25-26-27 間の角度を知ることができる。
( もちろん、各スクロールバー内から "residue" と "Z" を選択して、残基番号を入力しても "CZ" の二面角を得ることができる。 )
(Fig.9) 二面角と原子位置。

Fig.9 に示したように、 原子 29 と 46 が "シス" の位置にあるとき、この二面角は "0"°になる。
一方、それらが "トランス"の位置にあるとき、二面角は -180° (もしくは 180°) になる。
Fig.9 の視点 (= 30 → 31 方向の矢印 ) から見て、ベクトル 31-46 は 反時計方向に ある角度 (例えば 30° ) 回転すると、 二面角は 負 ( -30° ) になる。
一方、時計回りに回転すると、その角度は 正 になる。
(Fig.10) C-183 を挟む 原子間の距離。
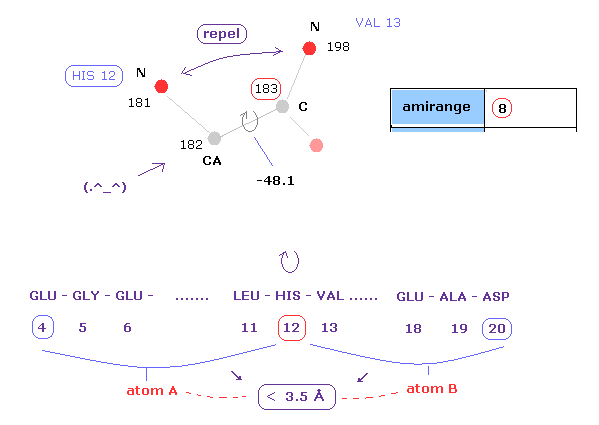
重要なのは 何が 二面角を決定するのか ということである。
ここでは 指定された二面角を挟んだ 両サイドの原子 ( A と B ) の間の 反発相互作用に注目した。
"8" の数値を "amirange" 横のテキストボックス内に入力すると、 このプログラムは 残基 "4" (= 12 - 8 ) と 残基 "20" (= 12+8) の間の 水素原子を除く すべての原子間距離を計算する。
1つの原子 A は 残基 4 から 12 に含まれ、 もう1つの原子 B は 残基 12 から 20 の間に含まれている。
ここで 炭素 182 (=CA ) と 183 (= C ) は この二面角に まったく影響を与えない。
( 例えば、182 と 198 の原子間距離は この二面角の回転で 変化しない。)
そのため このケースでは 原子 182 と 183 は この計算から除外されている。
(Fig.11) 182-183 の両サイドの2つの原子間距離が 3.5 Å 未満のもの。

このプログラムは コマンドプロンプト画面上に 水素原子を除いた
3.5 Å 未満の原子間距離のものをすべて表示させる。
このページに示したように、水素原子は 非常に弱い反発効果しか持たないため、ここでは この効果を無視できる。
例えば Fig.11 では、 酸素原子 (= プログラム番号 130 ) と 窒素 (= 198 ) の間の距離は 3.364 Å である。
この二面角に影響を与えうる 最も短い距離は 2.765 Å で、これは Fig.10 の 窒素 181 と 198 間の距離になる。
重要な点は 立体障害のために、ほとんどすべての原子間距離は
2.8-2.9 Å 以上になる。
( これは タンパク質構造を決定する際の重要な要因である。)
(Fig.12) C-183 後のアミノ酸すべてを 10°回転。
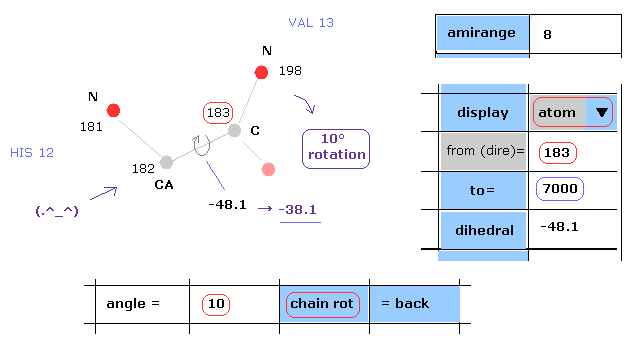
次に 炭素-183 の後のすべてのアミノ酸を 10°回転させる。
"183" と "7000" を "from(dire)=" と "to=" の横のテキストボックス内に入力する。
( ミオグロビンの最後の原子は 2843 であるため、この "7000" の数値は 2843 以上だったら何でもいい。 )
それから "angle=" の隣の テキストボックス内に "10" を入力して "chain rot" ボタンをクリックする。
このミオグロビンは 2843 原子を含んでいるため この場合では 炭素-183 後の すべての原子が 182-183 の原子間を軸として その周囲を 10°回転する。
( つまり この 二面角は -48.1°から -38.1°に変化する。 )
(Fig.13) C-183 後のすべての原子の 10°回転 - 残基指定。
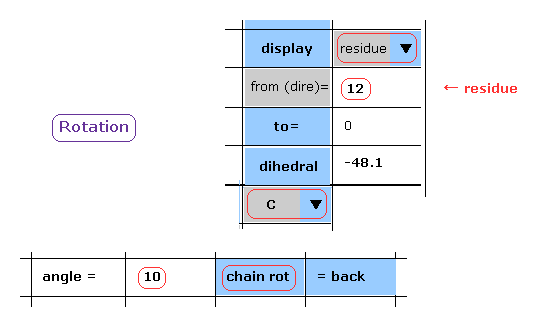
各スクロールバー内で "residue" と "C" を選択しても Fig.12 のように 回転させることができる。
"10" (= 回転角 ) と "12" (= 回転させる場所の 残基番号 ) を 各テキストボックス内に入力して "chain rot" ボタンをクリックする。
このケースでは、 "to" ボタン横の値に関わらず、C-183 移降の すべてのアミノ酸を 10°回転させる。
(Fig.14) 回転後の 原子間距離の変化。

コマンドプロンプト画面で、回転前より 短くなった 原子間距離が 画面に表示される。
Fig.14 に示したように、窒素 181 と 198 間の距離は 短くなりすぎる ( 2.765 Å → 2.690 Å )。
そのため この部分は 強い反発効果を発揮し、立体障害によって この回転を
ブロックする。
Fig.11 に示すように 回転前の 最も短い距離は 2.764 Å である。
そのため この最も短い長さが この回転によって ますます短くなってしまう。
(Fig.15) 回転戻す。 → -10°回転。
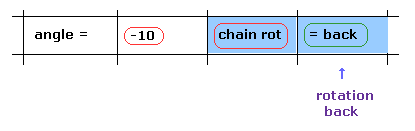
次に 逆方向への回転を調べる。
"=back" ボタンを押して 元の状態に戻る。
"-10" をテキストボックス内に入力して "chain rot" ボタンをクリックする。
このケースでは、炭素-183 後の すべてのアミノ酸が -10°回転する。
( Fig.9 に示したように、正の角度は時計回り、負の角度は 反時計回りである。)
(Fig.16) -10°回転。

Fig.16 に示したように、-10°回転においても、ある原子間距離は 短くなりすぎる。
酸素 149 と 窒素 214 間の距離は 2.698 Å となる。
このケースでは この部分のαへリックスが この回転によって きつくなりすぎてしまう。
つまり -48.1°の二面角が すべての障害物を避けた結果の 最も安定な状態であることが分かる。
これはつまり 1つ もしくは 2つの 際立った障害物が 各二面角を決定する キープレイヤーであることが分かる。
(Fig.17) 最も安定な二面角。

キーポイントは 最も短い原子間距離である。
なぜなら 例え 2,3つと言えども 際立って短い原子間距離が存在すれば、この回転は ブロックされ得るからである。
つまり、現在のタンパク質構造は すべての障害物を避けた結果であることを示している。
これが タンパク質構造を決定する 真の理由 と言える。
(Fig.18) ほぼすべての二面角は 固定されている。
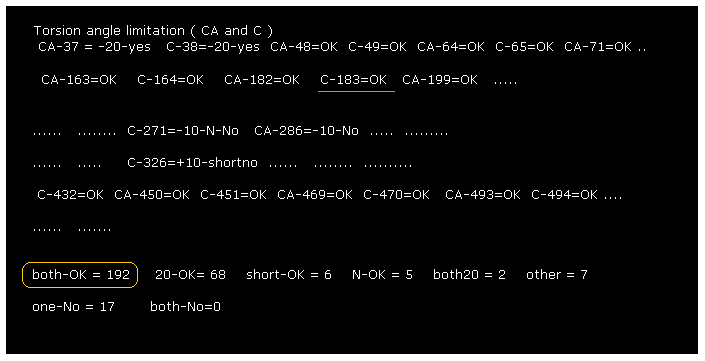
プログラム実行後、 "0" (= Rotate all angles, yes ) を入力すると、Fig.18 の結果が表示される。
"CA-37" は プログラム原子番号 "37" (= CA ) の場所における回転を意味している。
"both-OK" は Fig.17 に示したように 両方向 (= ±10° ) の回転が ブロックされており この状態が 安定であることを意味している。
"20-OK (yes)" は 1方向の回転が +10°でブロック、もう1方向の回転が -20°でブロックされていることを意味している。
つまり このケースでも 両方向の回転が ほぼ ブロックされて 安定であることを意味している。
"one-No" は 1方向の回転が ブロックされており、もう1方向の回転が
ブロックされていないことを意味している。
これらの部位は タンパク質の 表面近くにあることが多く、 おそらく この回転は 他のタンパク質か何かによって ブロックされていると思われる。
ミオグロビンにおける C,CA 部位で 両方向がブロックされている部分は トータルで 192箇所あり、 この数は 17 (= 1 方向がブロックされていない箇所)よりも はるかに大きい。
この結果は 1、2 の障害物 (= 最も短い原子間距離 ) でさえ 各二面角 を決定する際の 重要な要因となっていることを示唆している。
(Fig.18') 回転が 両方でブロックされている。
タンパク質全体の構造は 二面角 (= ねじれ角 ) によって決まる。
各回転可能部位では アミノ酸は ある程度は 自由に回転できる (= 回転異性体 )。
この部位に示したように、2つの離れたアミノ酸部位の 衝突が これらの回転を ブロックし、 タンパク質全体の構造を決定している要因でると言える。
つまり タンパク質は 回転する固体のように振る舞っていると言える。
(Fig.19) C 部位における回転。
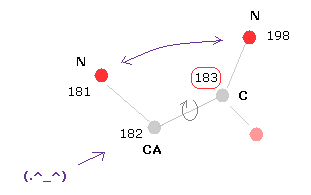
C 部位では、この C に近い 2つの窒素原子が 非常に重要である。
+10°方向回転によって この距離が さらに短くなり、これがブロックされる。
-10°回転では、この距離は 長くなるが、その代り 別の部分の原子同士が 互いに近づくため この回転はブロックされる。
このプログラムの計算は 最も短い原子間距離のみに注目している。
あるケースでは 元の N-N 長が -10°回転後も 最も短い距離のままのときもある。
これらのケースで この部位 N-N 長のみを除いて もし回転がブロックされた場合、これを -10-N-No と表示している。
ある部位では まれに 元の状態で 非常に短い ( 約 2.650 Å ) 原子間距離を含むケースがある。
この部位を除けば 回転がブロックされているとき、この部位を "+10-shortno" と表示している。
プログラム原子番号も表示されているので この部位を実際に回転させて これらの意味を確かめてほしい。
(Fig.20) アミノ酸計算部位の変更。

上のセクションでは Fig.10 に示すように ±8 に含まれるアミノ酸部位のみ計算した。
しかし アミノ酸番号が離れていても 実際の距離が近いケースもある。
そこで 上のプログラム内の 変数宣言を "int searchrange=20" のように変更する。
この変数は 計算するアミノ酸の範囲を意味している。
この値が 20 のとき、このプログラムは 回転部位を挟んだの両サイド ±20 アミノ酸以内の原子間距離を計算する。
( もしくは 下のプログラムを使うように。)
サンプルプログラム ( ami2.java. 計算範囲 ±20 )。
(Fig.21) 回転は ほぼすべてブロックされている (= both-OK ).
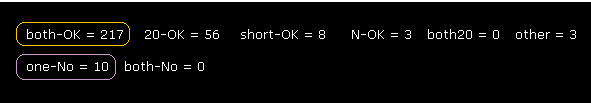
Fig.21 に示すように、計算部位を拡大すると、各二面角の回転が より顕著に ブロックされているのが分かる。
"both-OK" は 両方向の回転が ブロックされている という意味である。
( つまり ±10°の両方向の回転で 最も短い原子間距離が さらに短くなる。 = 両方向の回転がブロック。 )
両方向の回転が ブロックされた トータルの部位数 は 217 で、この数は 非ブロック部位 (= たった 10 ) の数より はるかに大きい。
(Fig.22) 2つの離れたアミノ酸部位が衝突。
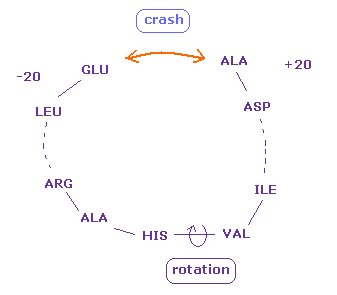
アミノ酸残基番号が離れていても 元の原子間距離が非常に短いときもある。
これらのケースでは そこから離れた部位の回転でも 2つの原子が Fig.22 に示したように衝突するケースもある。
結果的に Fig.18 の "192" (= ±8 の範囲 ) の数が Fig.21 で "217" に増加する。
これらの結果は タンパク質の構造は 固体のような性質を持つことを意味している。
つまり この固体の 一部分のみが衝突するだけで このコンフォーメーション変化は阻害されるということである。
非常に短い原子間距離が 少しあるだけで この部分は タンパク質構造において非常に重要な役割を果たしている。
(Fig.23) アラニンをフェニルアラニンに変更。
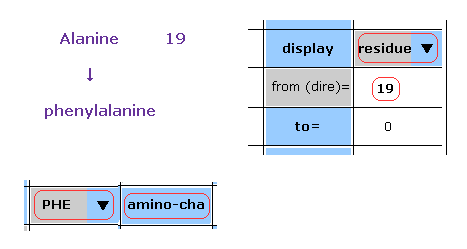
このプログラムでは、各アミノ酸の種類を変更できる。
例えば、残基 19 は アラニンである。
スクロールバー内から "residue" を選んで、 "19" を "from (dire)=" 横のテキストボックス内に入力する。
そして スクロールバー ( 7行目 ) 内 から "PHE" を選択して "amino-cha" ボタンをクリックする。
(Fig.24) アラニン → フェニルアラニン。
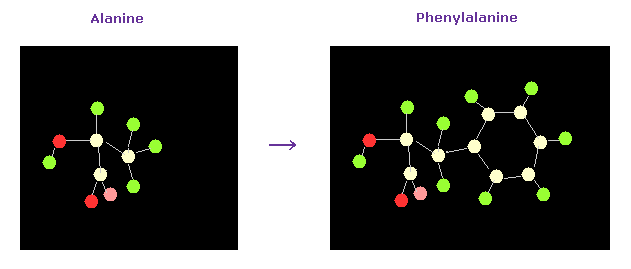
結果、指定のアミノ酸が変更された。
(Fig.25) 12 から 20 残基の 二面角 ( CB ) を変更。
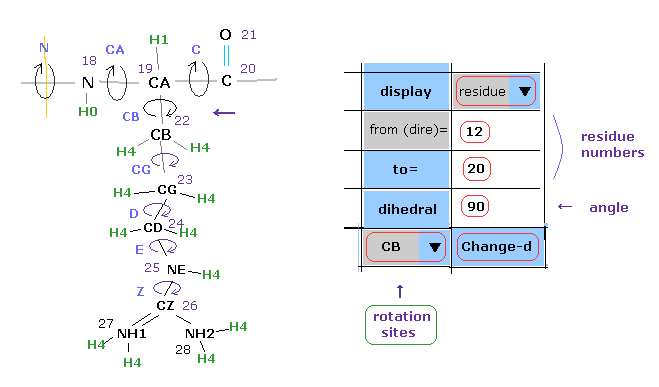
このプログラムでは、あるアミノ酸鎖に含まれる 任意の位置の 二面角を変更できる。
Fig.25 では、12 から 20 のアミノ酸に含まれる すべての CB の部位の二面角を変更している。
( 例えば GLY などの CB を含まないアミノ酸部分を含んでいるとき、これらのアミノ酸部分は 自動的にスキップされる。)
各スクロールバー内から "residue" と "CB" を選択して テキストボックス内に "12" と "20" の番号を入力し、"dihedral" ボタンの横に したい角度 "90" を入力する。
そして最後に "Change-d" ボタンをクリックする。
そうすると、12-20 アミノ酸に含まれる すべての CB 部分が90°に変化する。
この操作は "chain rot" ボタンとは 異なる。
"chain rot" では この回転は "=back" ボタンで戻るが "Change-d" では 元に戻らない。
(Fig.26) アミノ酸

このプログラムでは 新しいアミノ酸鎖を設計できる。
Fig.26 のように プログラム内の指定の場所を変更するように。
サンプル JAVA プログラム ( ami3.java タンパク質設計 )。
Fig.26 のように、 変数宣言を "int constr = 1" のように変更する。
"String sequence[]" は これから設計されるアミノ酸鎖配列である。
Fig.26 では 22 × ロイシンが作られる。.
変数 "resn" を アミノ酸の総数 (= "22" ) に変更するように。
"CAang" と "Cang" は CA と C すべての角度の初期値である。
CA と C の 二面角が -60° と -50°のとき、 このタンパク質は 典型的な αへリックスになる。
(Fig.27) αへリックスの水素結合 (= 全 22 アミノ酸 )。
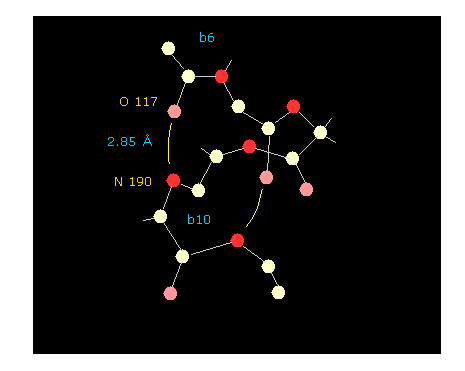
"hydrogen" と "chain" ボタンをクリックすると、水素原子なしの 主鎖のみ表示される。
ある 酸素 と 窒素 を続けてクリックすると、
その間の原子間距離が テキストボックス内に表示される。
Fig.27 に示すように、 残基 "6" の酸素と 残基 "10" の窒素が水素結合を形成し、この長さは 2.85 Å である。
もし 二面角をまとめて変更 ( "Change-d" ボタンを使う。 ) すると、この水素結合が 二面角に応じて どのように変化するかが分かる。一度試してみるといい。
(Fig.28) 窒素における 固定された相互作用部位。

このページで説明したとおり、この距離を計算するとき、Fig.28 のように 固定されたエリアを除いている。
例えば、Fig.28 の 窒素のケースのように、N と 線で囲まれた原子との原子間距離は C もしくは CA のアミノ酸鎖回転で変化しない。
これはつまり このエリア外の他の原子が 窒素近くの 各二面角を決定しているといえる。
(Fig.29) N-3 と N-4 の相互作用。

Fig.29 では、 窒素 3 と 4 ( N-3 と N-4 ) の距離が それらの間の二面角に影響を与えている。
一方で、 N-3 と CA ( もしくは C ) 間の 距離は この回転によって 変化しない。
つまり このケースでは、 Fig.28 に示したように CA や C の原子は 固定エリア内にあり、N-4 は その外にある。
(Fig.30) x-width の指定。
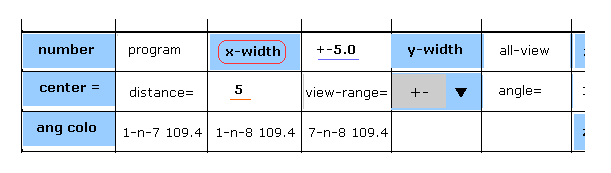
このプログラムでは、タンパク質全体の 断面図を見ることができる。
1例として x-z 平面 (= 左 )、 y-z 平面 (= 右 ) を 取り上げる。
( もちろん、任意の平面で この操作が有効である。)
Fig.30 で スクロールバー内から "+-" を選択して "5" (= 5.0 Å ) の数値を distance 横のテキストボックス内に入力して "x-width" ボタンをクリックする。
この状態では、Fig.31 に示したように x 方向へ 幅 5.0 Å の部分が y-z 平面上 (= 右画面 ) に表示される。
(Fig.31) x-width の指定。

スクロールバー内から "+" を選んでいれば 指定場所から x 方向に 0 から +5.0 Å の幅が表示される。
( "-" の場合は、 -5.0 Å から 0 の範囲となる。 )
注意: 例えば x-z 平面は 必ず x、z 方向の すべての原子を表示する。
そのため Fig.31 で "z width" を指定しても 画面は変化しない。
( なぜなら x-z と y-z の両方とも z 方向を含んでいるからである。)
上記のスクロールバー内で "all-view" を選んで、 x-width ( もしくは y, z- width ) ボタンをクリックすると、元の状態に戻れる。
"number" ボタンをクリックすると、下の原子画面に表示されている 原子番号の種類を変更できる。
基本的に 残基番号が短くて見やすい。
( また PDB 原子番号では、水素の番号が表示されておらず、これも見やすい。)
(Fig.32) テキストボックス内の表示原子を変更する。

"Atom" ボタンをクリックすると、 次の原子の列 ( 5 - 9 ) が表示される。
"+Z back-1" ボタンをクリックすると、1ページ前の列に戻る。
"+Y back" ボタンをクリックすると、 最初の原子の列 (= 0 - 4 ) に戻る。
"+X (A)" ボタンをクリックすると、選択した原子に対する相対座標が表示される。
"dis (A)" は 選択された原子と 各原子間の 距離である。
( このケースでは 0-N と 1-CA の距離は 1.473 Å である。 )
(Fig.33) 原子を "select" (= 選択 ) する。

スクロールバー内から条件 ("atom" もしくは "PDBat") を選んで、"select" ボタン横のテキストに 原子番号を入力して そのボタンをクリックする。
Fig.33 のケースでは、 プログラム原子番号 "40" (= トレオニンの炭素 CA ) が "選択原子" となる。 ( "*" のマークが付く。 )
"dis (A)" の列では、各原子と選択原子間の距離が表示される。
プログラム原子番号を用いて B サブユニット内の ある原子を指定するときは、原子番号に"10000" をつける。
( 例えば B サブユニット内の "18" の原子は "10018" で指定する。 )
PDB 原子番号は 各原子に特異的なため、この 10000 などは つける必要はない。
同様に、条件を選択後に 原子番号を "to" の横に入力して "to" ボタンをクリックすると、指定原子のテキストボックスへ ジュンプする。
" | " マークが "to" 原子に付加される。
この "to" 原子は 選択原子と異なり、単に テキストボックスへ移動するだけのものである。
Fig.33 で、 "PDBat" をスクロールバー内から選択していれば、"40"、"41" の代わりに "19" もしくは "20" を 各テキストボックス内に入力する。
( B サブユニット内では、プログラム原子番号に "10000" を つける必要がある。)
(Fig.34) 画面上の原子を直接クリックして "選択原子" を変更する。
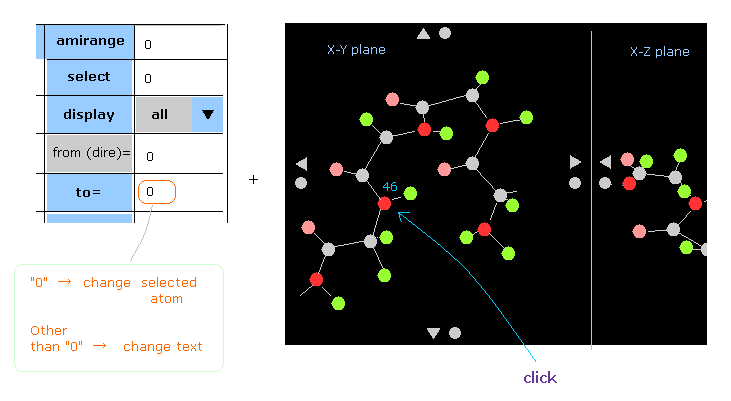
画面上の ある原子を直接クリックして 選択原子を変更できる。
例えば、"46" の赤の円 (= 窒素) をクリックする。
この時点で、"to=" 横のテキストボックス内に "0" が入力されている状態だと 選択原子が "46" に変更される。
( "to=" ボタンはクリックする必要はない。ただ その横に "0" を入力するだけでいい。 )
もし "to=" 横に "0" 以外の数値を入力した状態で "46" 原子の丸をクリックすると、選択原子は変更されないまま、"46" 原子のテキスト画面にジャンプする。
( つまりこのケースでは "*" のマークが "46" 原子につかない。)
(Fig.35) "ang colo", "zoom", "x-y,x-z" ボタン。
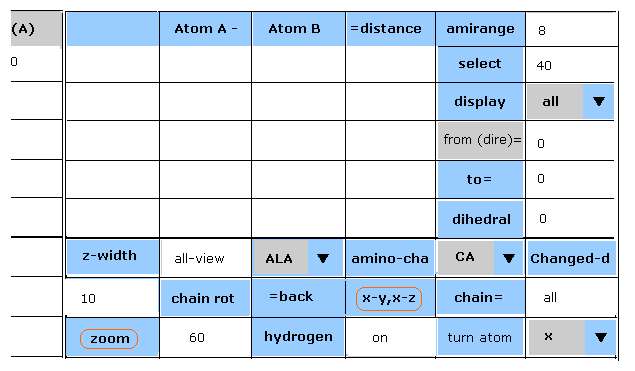
"zoom" ボタン横のテキストに数値を入力して そのボタンをクリックすると、タンパク質のズームが変化する。
"100" の値が最初の状態。
"200" を入力すると、 タンパク質全体が 2倍に拡大する。
"50" を入力すると、タンパク質全体が 元の像の半分に縮小する。
( 最初に タンパク質全体像を把握するために 縮小させるのがいい。)
"x-y、x-z" ボタンをクリックすると、左右の座標画面を変更できる。
最初が "x-y, x-z" 平面、 2番目が "x-y, y-z" 平面、 3番目が "x-z,y-z" 平面である。
"ang colo" ボタンをクリックすると ( Fig.30 参照 )、サブユニットA が 赤色、 B が 緑色、 C がピンク、 D が黄色になる。
( また HETATM 原子は 紫になる。 )
そのため A と B のタンパク質サブユニットを容易に区別し その相互作用構造を把握できる。
(Fig.36) "center" ボタン。

ある値 ( 例えば "5" ) を "distance" 横の テキストボックス内に入力して"center" ボタンをクリックすると、選択原子から 5.0 Å 以内の距離の原子のみ画面に表示される。
初期値は 80 Å である。
この "center" と "width" ボタンを組み合わせれば、タンパク質の様々な部分の構造が 容易に理解できる。
(Fig.37) 2つの任意の原子間の距離。
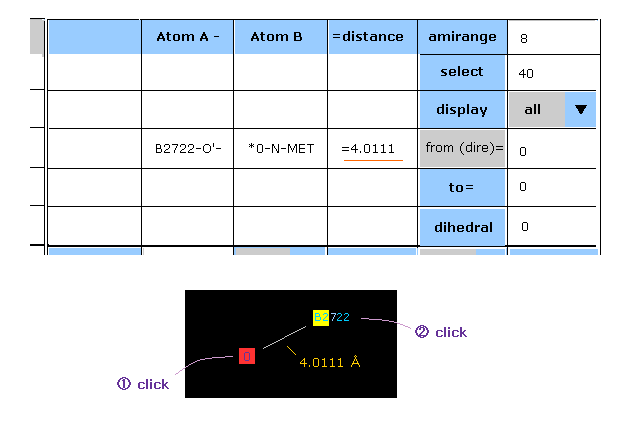
2つの任意の原子を続けてクリックすると、それらの原子間距離がテキストボックス内に表示される。
Fig.37 では、最初に メチオニンの N 原子 (= 原子番号 0 ) をクリックし、次に H2O の酸素原子 (= B サブユニット 2722 ) をクリックしている。
これらの数値はプログラム原子番号である。
Fig.37 のケースでは、O の原子が選択されており、それが 四角形になっている。
また直前にクリックした原子は 黄色の四角形になる。
( もちろん、"dis A" の列を見ても、各原子と選択原子間の距離を知ることができる。)
他のコマンドは ほぼ このページと同じである。
"from-to-display" コマンドでは スクロールバー内から "residue" の条件のみ選択できる。
例えば、"from" と "to" の横のテキストに "1" と "5" を入力して "display" ボタンをクリックすると、
サブユニット A の 残基番号 1-5 のアミノ酸のみ画面に表示される。
"10001" と "10005" をテキストボックス内 ( "from" と "to" の横 ) に入力して "display" ボタンをクリックすると、サブユニットB の残基番号 1-5 のアミノ酸のみが 画面上に表示される。
"C" と "D" のサブユニットに関しては、"20000" もしくは "30000" を 残基番号に付加する。
"dihedral"、 "chain rot" ボタンに関しては、 このページ とほぼ同じである。
サブユニット B 内の 二面角を知りたいときは、 "10000" を プログラム原子番号に付加する。
( PDB 原子番号に "10000" をつける必要はない。 )
また このプログラムでは 二面角の部位を Fig.13 に示したように residue + 部位の スクロールバーでも指定できる。
基本的に 1つのタンパク質サブユニットは "TER" で終わる。
"TER" に続くアミノ酸は 次のサブユニットとなる。
"HETATM" の部分は 直前のサブユニットに吸収されてカウントされる。
(Fig.38) タンパク質全体を ある方向へ回転させる。
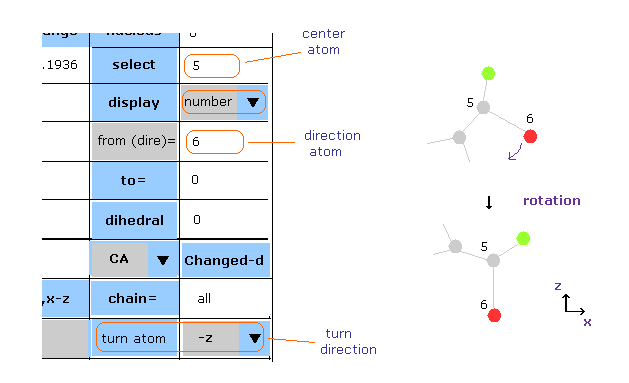
最初に ある原子を選択 (= select ) する ( これが回転中心になる )。
次に スクロールバー内から "atom" もしくは "PDBat" を選択した後、 " from (dire)= " の横のテキストボックス内に ある原子番号を入力して 下のスクロールバー内から 方向 ( x, y, z, -x, -y, -z ) を選択後、"turn atom" ボタンをクリックする。
Fig.38 のケースでは、 ベクトル 5 → 6 原子 を -z 方向へ向ける。
もちろん、B サブユニット内 ( PDB 原子番号を使わず ) の原子を選択するときは、プログラム原子番号に "10000" を付ける必要がある。
( つまり B サブユニットの プログラム原子番号 "1" は "10001" で指定する。 )

2013/9/1 updated This site is link free.