Top page (correct Bohr model including the two-electron atoms)
Back to multi-atomic molecules page
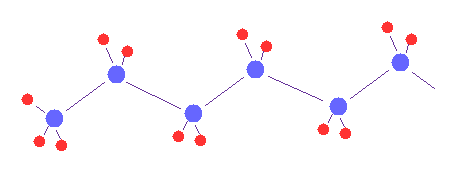
(Fig.1) Multi-atomic molecules.
Using the sample program below, we can visualize and compute various multi-atomic molecules, if we change initial data.
( The method of changing initial data is explained later. )
First, we think about various methane-like, H2O-like and formaldehyde-like molecules.
Sample JAVA program to compute various molecules.
If you display source code of the upper program page, you can save and compile it as it is.
( You can copy and paste, too. But this program is a little long. )
This class name is "orbit", So save this text editor as "orbit.java", and compile it.
If you see notes such as "Recompile with -Xlint:unchecked for detailes", you can neglect those messages, and run it as it is.
(Fig.2) Choosing methane molecule (= CH4 ).
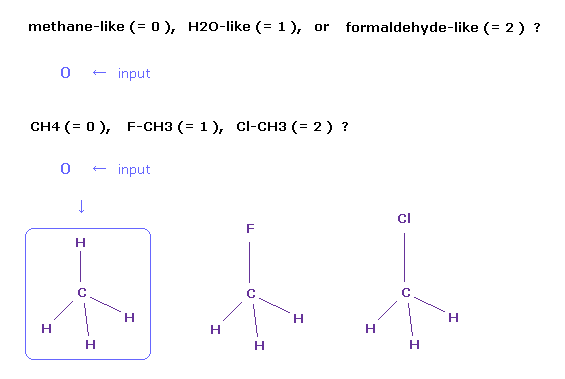
After running the program, a sentence of " methane-like (= 0 ), H2O-like (= 1 ), formaldehyde-like (= 2 ) " is displayed on the screen.
If you input "0" and press the Enter key, " CH4 (= 0 ), F-CH3 (= 1 ), Cl-CH3 (= 2 ) " is displayed next.
If you input "0" and press Enter key, methane (= CH4 ) is showed on the screen.
Other methane-like molecules are fluoromethane (= F-CH3 ) and chloromethane (= Cl-CH3 ), and you input "1" or "2", respectively, in these cases.
(Fig.3) Choosing OFH (= hydrogen-fluoro-oxygen ) molecule.
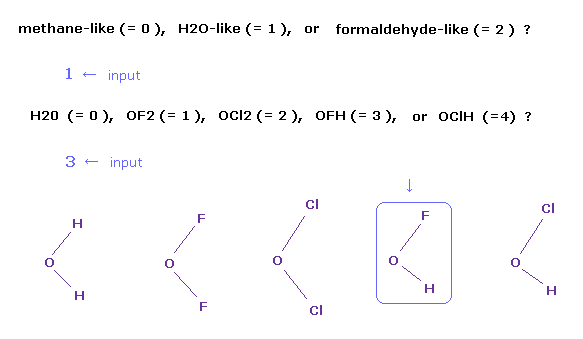
After running the program, a sentence of " methane-like (= 0 ), H2O-like (= 1 ), formaldehyde-like (= 2 ) " is displayed on the screen.
If you input "1" and press the Enter key, which means H2O-like molecules.
So " H2O (= 0 ), OF2 (= 1 ), OCl2 (= 2 ), OFH (= 3 ), or OClH (= 4 ) ? " is displayed next.
If you input "3" and press Enter key, OFH molecule is showed on the screen.
In case of formaldehyde-like (= 2 ), you can choose formaldehyde ( O=CH2 ), O=CF2, or O=CCl2.
(Fig.4) fluoromethane (= F-CH3 ) molecule.
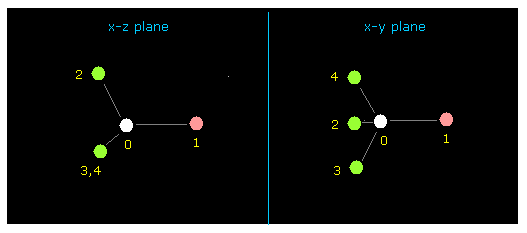
After running above program, input "0" (= methane-lile ), and then "1" (= F-CH3 ).
Cross sections ( x-z and x-y planes ) of F-CH3 is displayed on the screen.
This screen is "overall molecule", in which abosolute coordinates of each atom is showed on the textboxes ( +X, +Y, +Z ).
( unit: 1 MM = 1.0 × 10-14 meter ).
Fluorine (= atom 1 ) is displayed as pink circle, and hydrogen (= atom 2, 3, 4 ) is green, carbon (= atom 0 ) is white circles.
(Fig.4') fluoromethane (= F-CH3 ) molecule.
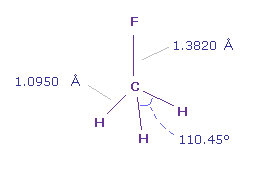
C-F bond length is 1.3820 Å and C-H bond length is 1.0950 Å.
On the screen, fluorine (= F ) atom is on x axis, and "nuc" in the textboxes means lengths between each atom and 0 (= C ) atom.
Actual H-F-H angle is 110.45 degree, but it's almost 109.45 degree of tetrahedron.
So here we think about usual 109.45 degree. ( Of course, you can change these coordinates, as I explain later. )
These bond angles ( degree ) are showed in the last line of "angle".
For example, "1-n-2" means the angle of atom 1 (= F ) - C nuc - atom 2 (= H )
(Fig.5) Force = 1000.
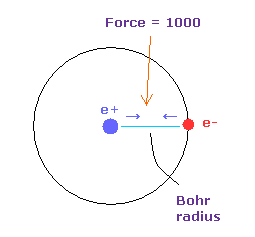
( FX, FY, FZ ) on the textboxes means force component acting on each nucleus.
For each nucleus to be stable, these values need to be close to zero.
Force unit is shown in Fig.5.
The force between electron and nucleus in H atom ground state is supposed to be "1000".
So when the distance between electron and +e nucleus is Bohr radius, its force becomes 1000.
For example the force of 1468 is 1.468 times that in H atom ground state.
"tV" means total potential energy of F-CH3 molecule.
And "V-1" means total potential energy of two atomic ( C and F = 1 ) molecules.
This "overall molecule" screen is for much larger molecule, so in case of small F-CH3, we don't use this screen so often.
(Fig.6) Each nucleus and its valence electrons.
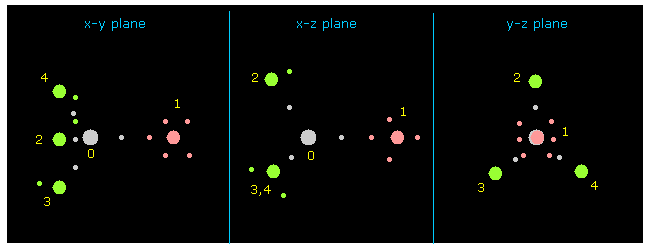
Next click "nucleus" button at upper right.
"0" is chosen inside the textbox next to "nucleus" button, so 0 atom (= C ) is dispayed at the center on each x-y, x-z and y-z planes.
Other atoms and its valence electrons (= small circles ) are displayed around C nucleus.
( FX, FY, FZ ) means force component acting on each nucleus, and "cf" is force component toward C nucleus.
( If this "cf" is positive, its atom is attracted toward C nucleus. )
If you input some values on ( +X, +Y, +Z ) textboxes, and press enter key, you can change relative coordinates of each atoms.
(Fig.7) Atom 1 is turned toward y axis.
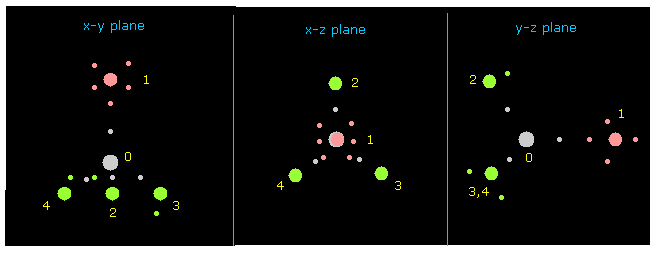
Next input "1" on the textbox next to "direction" button, and click "direction" button.
This button rotates overall molecules, and turn the designated atom ( in this case, atom 1 = F ) toward y axis.
Besides this button, you can rotate overall molecules clicking "x-y ang", "x-z ang", or "y-z ang" buttons.
Input some angle's value (= degree ), and click these buttons.
If so, overall molecule is rotated on each plane by the designated angle.
(Fig.8) Fluorine picture (= atom 1 ) .
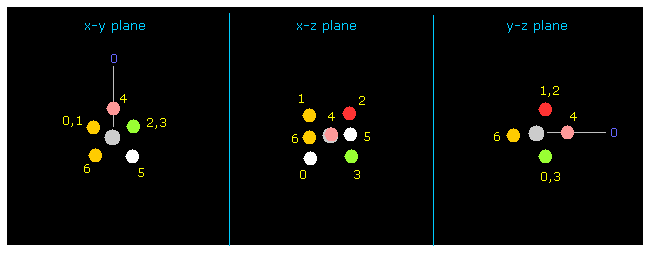
Next input "1" on the textbox next to "electron" button, and click "electron" button.
If so, atom 1 (= fluorine in this case ) and its 7 valence electrons are displayed on the screen.
Carbon (= atom 0 ) is turned toward y axis only in this electron picture.
If you click "nucleus" or "top nucl" buttons, you can go back to Fig.7 (= each nucleus picture ) or
overall molecule picture of first section again.
You can change each relative coordinate of valence electrons by inputing arbitrary values (MM) into textboxes (+X, +Y, +Z) and press enter key.
Other parameters' meanings are almost same as this page.
(Fig.9) Rotate fluorine by 90 degree on x-y plane.
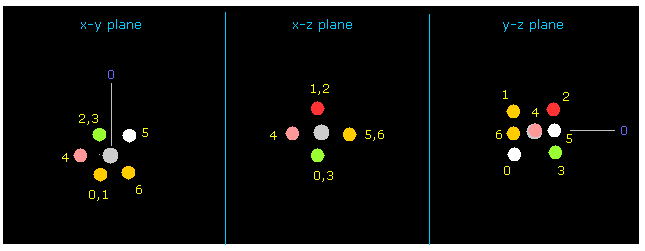
As shown in Fig.7 and Fig.8, fluorine valence electron "4" in this first picture just points to carbon.
So the repulsive force between this electron "4" and one of carbon valence electron is too strong, and makes this molecule unstable.
Input 90 degree into the textbox next to "x-y ang" buton, and click "x-y ang" button.
Then fluorine is rotated by 90 degrees on x-y plane, and valence electrons of fluoine and carbon can avoid each other (= Fig.9, 10 ).
(Fig.10) Rotate fluorine by 90 degree on x-y plane to avoid repulsive force.
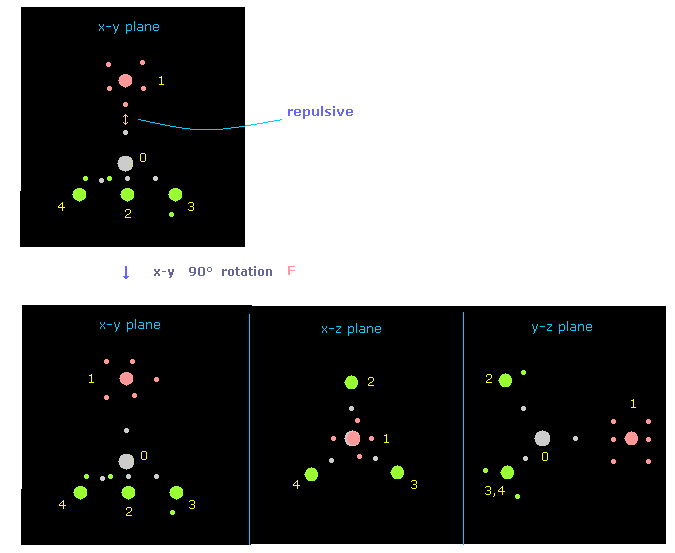
If you input "0" on the textbox next to "nucleus", and click "nucleus" button, you can understand only fluorine is rotated by 90 degree on x-y plane, from the viewpoint of carbon and its four neighboring atoms.
( Caution: plus angle is counterclockwise rotation in electron picture, so in this case, atomic picture is rotated in the opposite direction. )
"nucleus" button displays some atom (= input its number on the designated textbox) and its neighboring atoms.
And "top nucl" button displays overall molecule, centering some atom designated on textbox.
( In small atoms such as CH4 and F-CH4, these "nucleus" and "top nucl" button give almost same picture. )
(Fig.11) Force acting on C nucleus is "balanced".
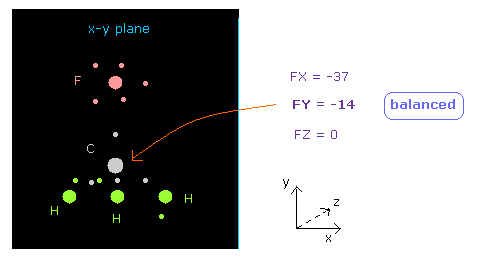
See "now 0" line below in nucleus picture, which means atom 0 (= carbon ) is at the center.
Textboxes next to "now 0", force acting on this carbon atom is displayed.
You can see FY = -14, which is almost zero.
So force acting on central carbon nucleus is just balanced among F and 3× H atoms, in this case.
If you input "12000" on the textbox of "+Y (MM)" in "atom 1" line, and press enter key, the bond length F-C changes to 12000 MM (= 1.2000 Å ).
In this case, force acting on carbon becomes ( FY = -125 ), due to stronger repulsive force by F nucleus.
So the bond length of 13820 MM (= 1.3820 Å ) is balanced one.
(Fig.12) Force acting on four valence electrons are "balanced".
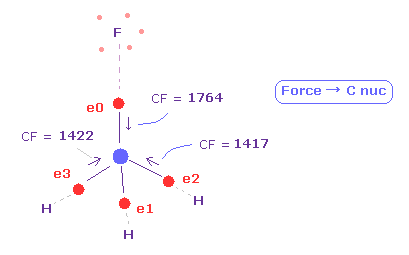
Again input "13820" on the textbox of "+Y (MM)" in "atom 1" line, and press enter key.
Next input "0" next to "electron" button, anc click "electron" button.
Then four valence electrons ( 0-3 ) of carbon atom is displayed.
( Blue number around them represents atomic number around. )
See force "CF" of each electron, which means force component toward C nucleus acting on each valence electron.
As shown in Fig.12, when F-C and H-C length is 1.3820 Å and 1.0950 Å, these forces are almost equal to each other.
CF = 1764 at F side, and CF = 1417 at H side.
(Fig.13) Force balance is broken.
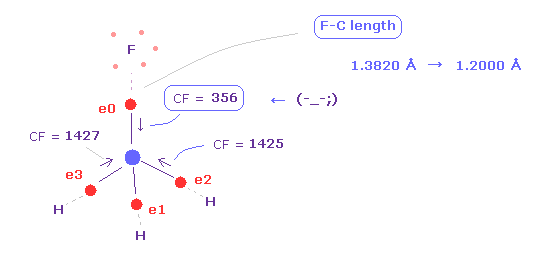
Again click "nucleus" button to atomic picture, and change +Y coordinate to "12000" from "13820".
This means F-C bond length is shorter ( 1.3820 Å → 1.2000 Å ).
Then click "electron" button ( neighboring text is "0" ), and see force CF of carbon valence electrons again.
As you see in Fig.13, when F-C length is shorter (= 1.2000 Å ), carbon valence electron "0" is too attracted to fluorine nucleus.
So CF = 356 is too weak, comparing with other CF = 1425.
( Of course, fluorine nucleus is also attracted to the electron too much. )
This means these particles become unstable.
As shown in this page, these unstability of valence electron and nucleus determines bond length such as C-H = about 1.0900 Å
Actually if you make C-H bond length shorter to 0.9500 Å from 1.0950 Å, CF of carbon valence electron at this H side becomes negative = -118.
( Of course, in this strange case, carobon valence electron is attracted to H nucleus instead of carbon ! )
As I said above, there are three patterns of screen.
Click "nucleus" (= each nucleus and its neighboring atoms ), "electron" (= each atom and its valence electrons ), or "top nucl" (= overall molecule ).
(Fig.14) Electron picture (= F ) .
In "electron" picture, "nuc" means the distance between each electron and its nucleus.
You can change this "nuc" by inputting some value on the designated textbox and "nuc" button.
Central charge Z can be changed, when you input some value, and click "charge Z" button.
If you click "inversion" button, all valence electrons showed on the screen move to just opposite positions of nucleus.
If you choose some atomic kind in the scroll bar, and click "A atom" button, you can change the kind of atom ( H, C, N, O, F, Si, P, S, Cl ).
When you want to adjust valence electron's position, use "x-y ang", "x-z ang", and "y-z ang" button, and rotate them in the proper positions.
Or you can change relative coordinates on textboxes freely.
(Fig.15) Hydrogen atom's type 0, 1, 2, or 3.
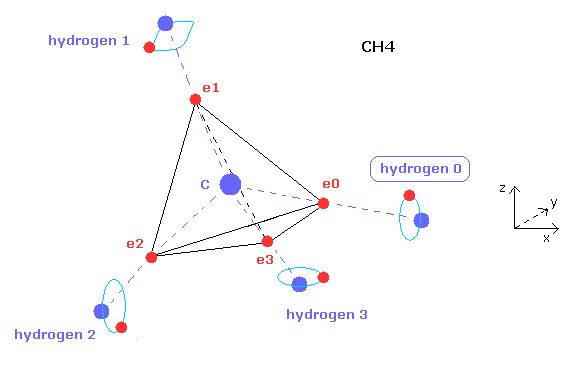
After clicking "electron" button and displaying some hydrogen electron, if you input the value of 0, 1, 2, or 3 on the designated textbox and click "hydrogen" button, you can change the type of hydrogne atom.
Hydrogen has only one electron, so we need to adjust their symmetric position.
For example, in methane (CH4), atom 1 is hydrogen 0, atom 2,3,4 are hydrogen 1,2,3, respectively.
You can manipulate only hydrogen 0 electron's position, and then electrons of hydrogen 1,2,3 are automatically arranged symmetrically based on hydrogen 0 electron.
You need to determine hydrogen-type based on the structure of Fig.15.
For example, in fluoromethane, atom 1 is fluorine, atom 2,3,4 are hydrogen 0,1,3 respectively.
In electron's picture, number of de Broglie waves are shown in "Waves" column.
In multi-electron atoms except for hydrogen, "waves" is computed only inside its atom.
( For example, in carbon, forces from surrounding atoms are almost balanced, so "waves" need to be computed inside only carbon. )
And "direw" means de Broglie waves, considering all direct forces acting on each electron.
In hydrogen atom, these "direw" and "Waves" are the same.
"dia" and "ave" means the average de Broglie waves of all valence electrons in "direw" and "Waves".
In hydrogen atom, we compute the electron after moving by force (= second line ).
These methods are completely same as this page.
(Fig.16) Multi-atomic molecules.
Sample JAVA program to compute various molecules.
Like the program above, you can compile the text as it is.
( Save this text as "orbitt.java", and compile it. )
You need to manipulate atomic parameters inside
"--- You can change freely initial data -----".
(Fig.17) Multi-atomic molecules.

First, you need to input total atom's number into "maxa".
For example, O=CF2 is "4", H2O is "3".
And you need to prepare the arrays of the same number as maxa.
( 0, 1, 2 ) component is absolute coordinate (= x, y, z ).
"3" component is central positive charge ( H=1, C=4.22, N=5.25, O=6.273, F=7.3, Si=5.05, P=6.12, S=7.18, Cl=8.26 ).
"4" component is electron's radius, unit of MM ( H = 4500, C=6415, O=4596, N=5332, F=4003, Si=11500, S=8622, P=9825, Cl=7640 ).
See also this page.
In model2, "0" component is number of valence electrons ( H=1, C=4, O=6, F=7 ...).
"1-6" component is neighboring atomic number.
In this case, neighboring atom of carbon (= atom 0 ) is oxygen (= atom 1), two F (= atom 2, 3 ).
And you need to input "-1" in all other components of "1-7".
The last "8" component means hydrogen type 0-3.
For example, in CH4, atom 1-4 is hydrogen 0-3, and you can manipulate only hydrogen 0's electron coordinate, and hydrogens 1-3's electrons are automatically and symmetrically changed based on hydrogen 0.
If you input some atomic nucmber, and click "top nul" button, overall molecule is displayed with the designated atom at their center.
Input some values (MM) on textboxes ( x, y, z from the left ) next to "move" button , and click "move" button, you can move overall molecule.
If you input "overall" button, you can change between "overall" and "each at".
In "each at" state, when you input some atomic number below "each at" text, and click "move" button, only designated atom moves.

2013/5/10 updated. Feel free to link to this site.