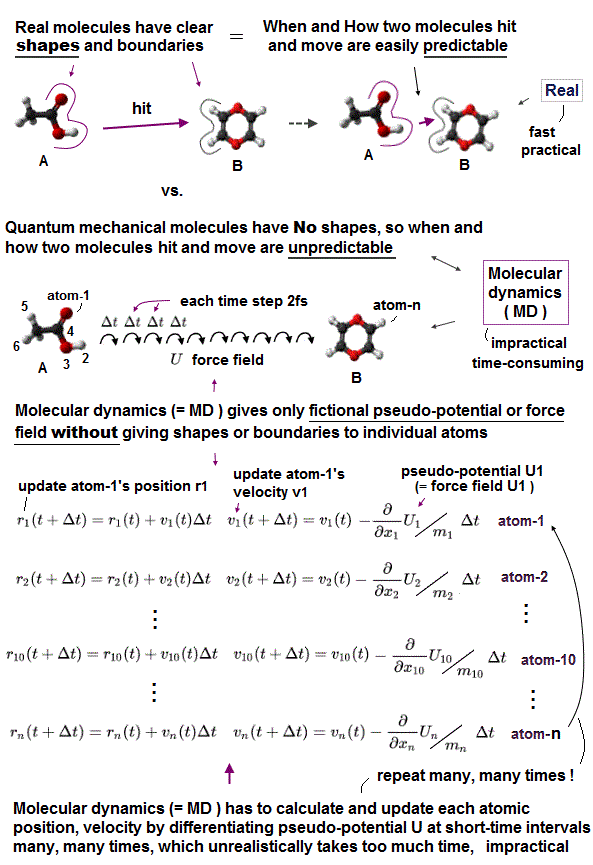
(Fig.1) Treating molecules as real objects with shape can easily predict molecular motion. ← The unrealistically time-consuming MD or quantum mechanics is unnecessary, just hampering technology.
Today's AI, Alphafold, protein docking simulating methods based on static protein structure database can deal with only useless static proteins ( this 4th-paragraph, this p.3-last ), with bad prediction rates ( this p.12-2nd-paragraph, this p.4-right ).
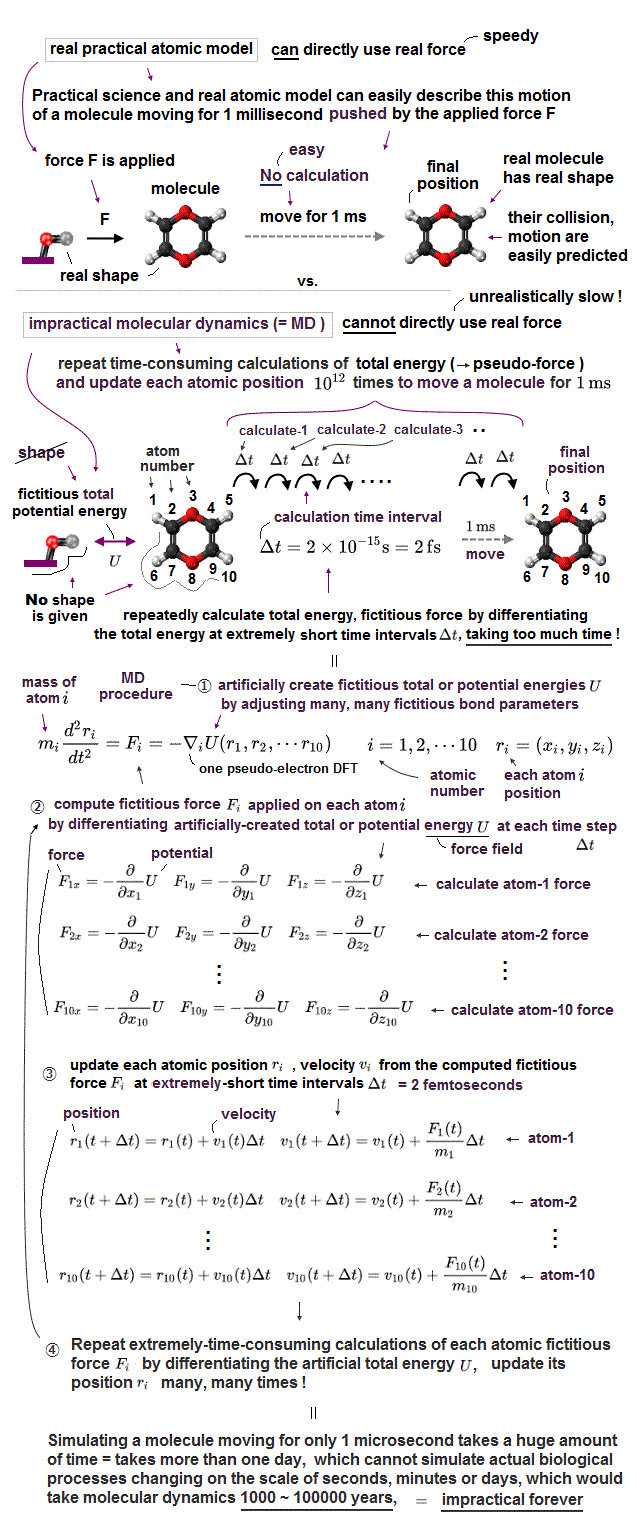
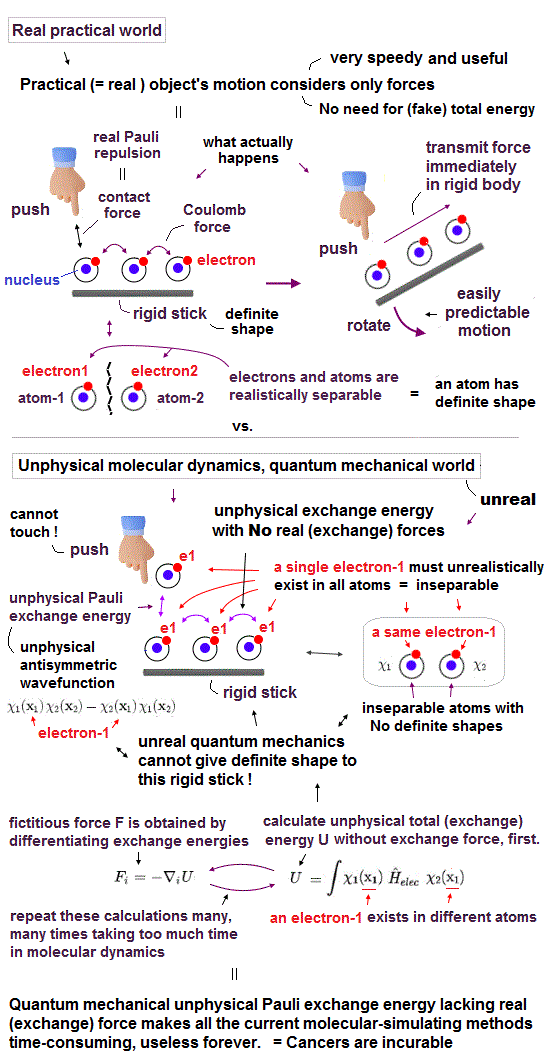
Due to the unphysical quantum mechanics unable to give real shapes to individual atoms, today's only molecular motion simulating method called molecular dynamics (= MD ), which is useless and extremely time-consuming ( this 9~11th-paragraphs ), also cannot define concrete shapes of individual molecules.
The unphysical quantum mechanics or molecular dynamics (= MD ) cannot even treat a rigid molecule as a real rigid object with shape.
Instead, MD has to move positions of all shapeless atoms inside a molecule individually (= take too much time ! ), little by little, based on pseudo-potential U called force field at short time intervals (= each time interval must be less then 2fs = 2×10-15 s ), repeatedly, many, many times, which takes unrealistically too much time.
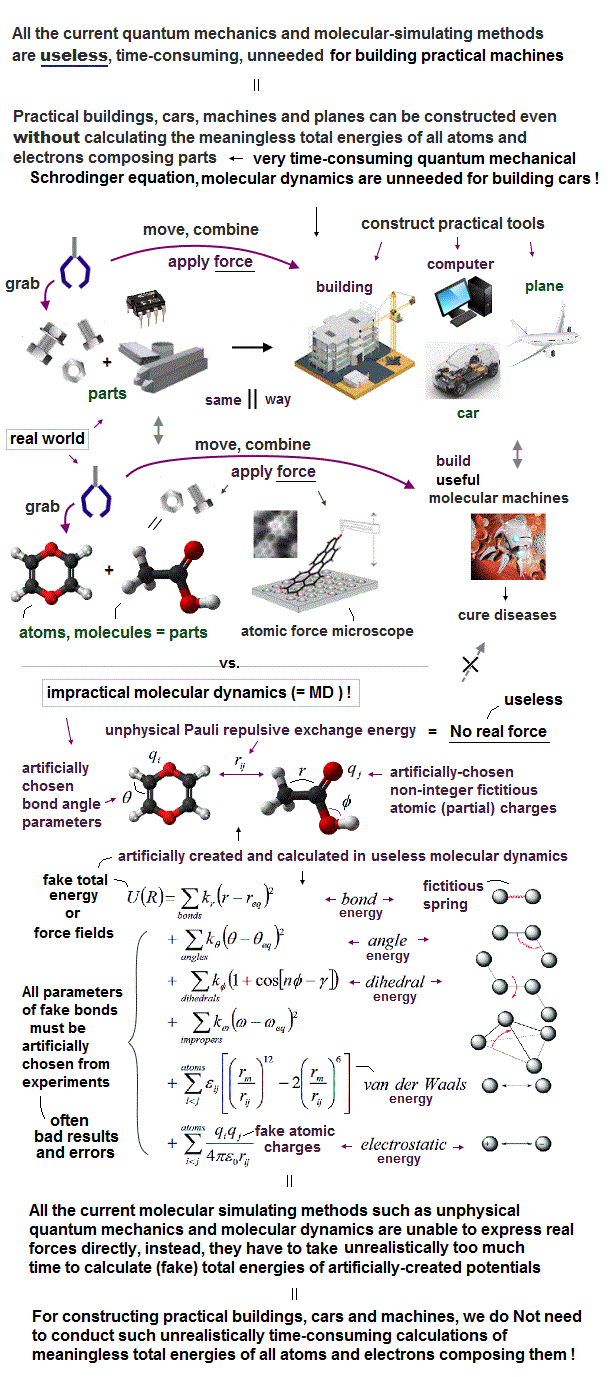
(Fig.2) MD pseudo-potential or force field U's parameters must be artificially chosen.
Molecular dynamics (= MD ) has to update positions of all shapeless atoms (inside a molecule) individually (= instead of moving a rigid molecule as a real rigid object with shape ), little by little, based on pseudo-forces caused by differentiating the force-field potential U ( this p.17~36 ) at short time intervals (= each time step must be shorter than 2 fs = 2 ×: 10-15 s ) repeatedly, many, many times ( this or this p.5-19, this p.1-14, this p.2-4, this p.2-3 ), which takes too much time, hampering science.
This-p.8-left-last-paragraph says -- Useless artificial MD
"MD simulations require significant computational resources and time, and
the choice of simulation parameters and the accuracy of the force
field can directly affect the reliability of the simulation results."
This-3. says -- Empirical force field
"The accuracy of MD simulations depends on the quality of the force field (potential energy function) used. Force fields are models that approximate the interactions between atoms. Their accuracy varies, and some force fields may not capture specific interactions accurately. Note that force fields are empirical models, meaning they are designed based on experimental data."
↑ MD is useless, too time-consuming, and cannot predict annything due to artificial choice of force-field potential's parameters.
This MD's force field potential's parameters must be artificially determined based on experimental results, empirically, instead of useless quantum mechanical prediction.
This-p.2-1st-paragraph says -- Useless quantum mechanics
"It is Not yet feasible to calculate directly such surfaces for
macromolecules with high accuracy by means of quantum chemistry
electronic structure calculations, so most practical simulations use a set
of simple classical functions to represent the energy, adjusting a
large number of parameters to optimize agreement with experimental
data"
This-p.1-right-1st-paragraph says -- No quantum mechanics
"it is difficult to obtain interatomic repulsion potential or reliable
dispersion energy from the first principles (= quantum mechaniccal prediction is useless )"
This-Why is the force field important ?-2nd-paragaph says -- Artificial choice
"Different force fields may have different assumptions and approximations, which may affect how well they represent the physical and chemical properties of your system. For example, some force fields may be more suitable for proteins than for nucleic acids"
↑ No universally-correct force field potential.
Choosing inappropriate parameters cause wrong results ( this p.5-limitation, this p.2-left, ), so molecular dynamics (= MD ) unable to predict any physical values or molecular behavior is useless, just impractically time-consuming ( this p.2-1st-paragraph ).
This p.2-2nd-paragraph says -- Artificial parameters
"even small errors
in the force field parameters can lead to catastrophic failure in long-time simulation – ML (= machine learning ) force fields may
exhibit pathological behavior,.. causing the simulation to become unstable or explode"
This-p.1-last-paragraph (2023) says -- Wrong MD simulation
"However, experimental data and simulation results often show discrepancies. One of the main reasons
for this is that the all-atom force fields (FFs) such as AMBER, CHARMM..
were developed to reproduce the properties of stable globular proteins"
Some force field potentials may be adjusted based on quantum mechanical DFT's pseudo-potential (= quantum mechanics or DFT treating the whole molecule as one pseudo-electron model just choosing exchange-correlation pseudo-potentials is also empirical, unable to predict any values, so fake ab-initio ), which is also useless too time-consuming and often giving wrong results ( this p.2-left-3rd-paragraph ).
This paper ↓ -- Useless quantum mechanics
p.2-left-IV. says
"However DFT’s
precision strongly depends on the choice of the functional" ← Quantum mechanical mainstream DFT cannot predict anything.
p.4-left-2nd-paragraph says --
"We employed the ωB97M-D3BJ functional (= empirically-adjusted functional = exchange potential, this-p.1-left-last, so No quantum mechanical prediction )"
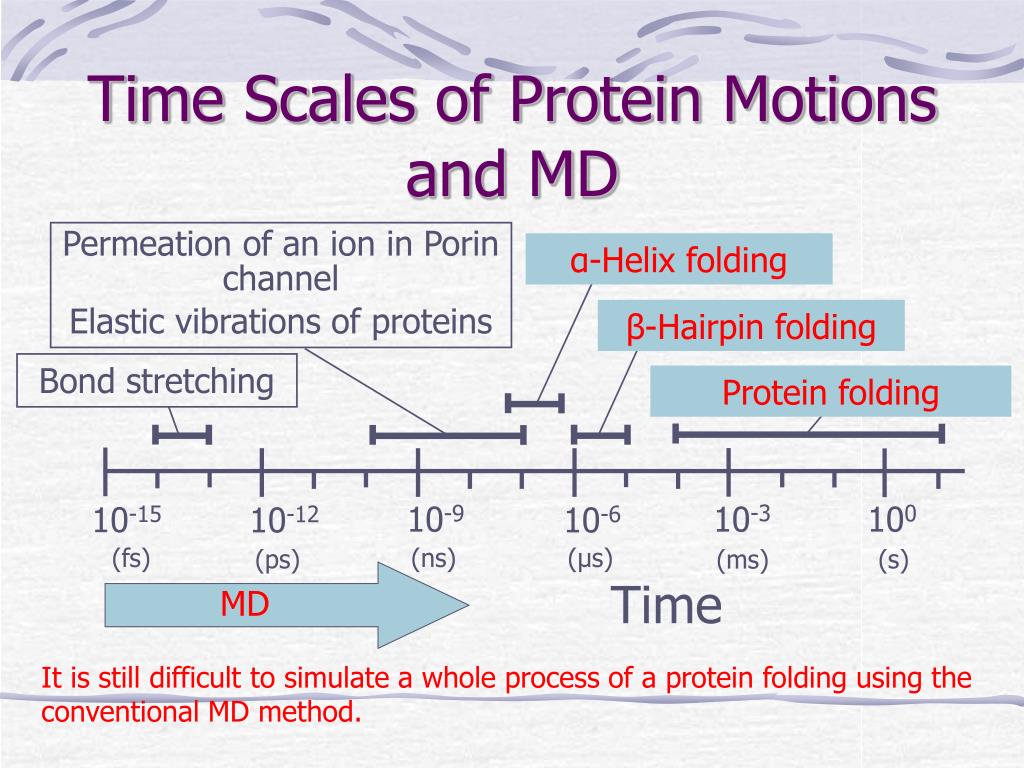
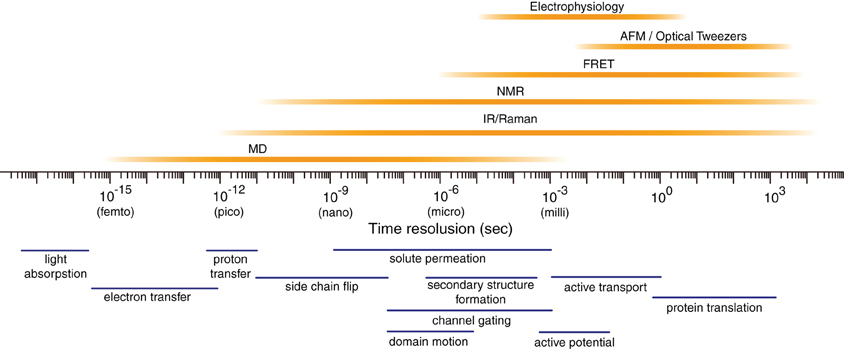
(Fig.3) The time-consuming molecular dynamics cannot simulate seconds ~ hours enzymatic reactions or protein's folding. ← Drug discovery is impossible.
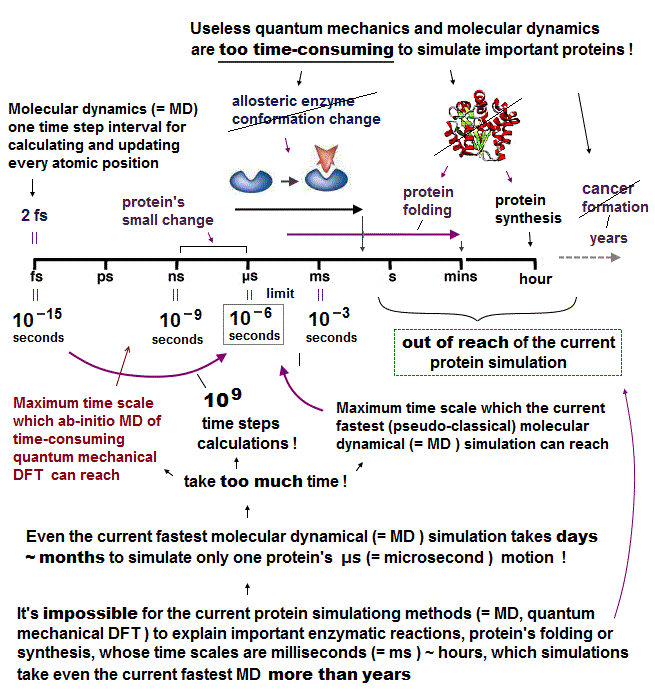
In molecular dynamics (= MD ) whose one time step (= time interval Δt ) must be less than 2fs (= 10-15 s, this p.5 ), simulation of just 1 μs (= 10-6 s ) molecular motion needs to calculate as many as 109 time steps repeatedly ( 1μs/1fs = 109 ), which takes impractically too much time (= more than days, this 3rd~4th-paragraphs ).
This-lower current limitation says -- Too time-consuming MD
"one-microsecond simulation of a relatively small system (estimated to be 25,000 atoms) running on 24 processors, which can take several months to complete ( this p.3-right-1st-paragraph )."
In most molecules or proteins, the time-consuming MD cannot simulate more than 1 μs motion. ← Even just nano-second (= ns ) protein simulation takes too much time ( this-lower, this-p.13 table.5 ).
So today's only molecular simulating method = MD cannot simulate important biological (enzymatic) reactions, proteins' folding ( this p.1-right-1st-paragraph ), synthesis, translation, which biological processes happen in milliseconds ~ minutes ~ hours ( this p.2-left-last~right ) time scale.
This p.2-left-1st-paragraph says -- Impractical MD
"folding times range from
microseconds to tens of minutes.... simulating the folding of
these larger slower folding proteins using standard molecular
dynamics (MD) simulations will remain out of reach for the
foreseeable future"
This p.3-left-2nd-paragraph says -- Too time-consuming MD
"atomistic MD
calculations.. being able to
compute few nanoseconds with a personal computer, but far from
the millisecond to second timescales of domain motions and
allosteric transitions occurring in some enzymes"
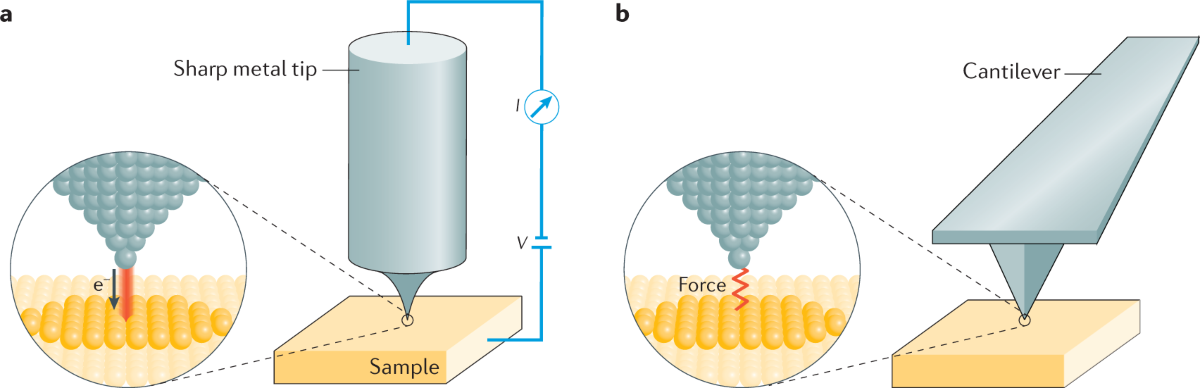
(Fig.4) The unphysical quantum mechanical shapeless atoms and extremely-time-consuming MD obstructs nano-technology.
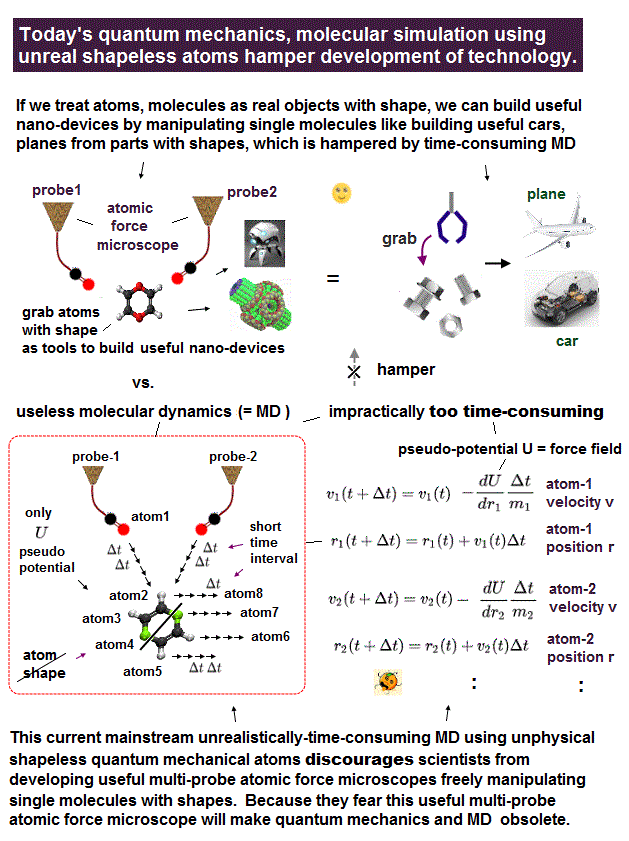
This current only molecular simulating method = unrealistically-time-consuming molecular dynamics (= MD ) is the main culprit of hampering today's nano-technology by preventing developing useful multi-probe atomic force microscopes..
To build practical cars, machines, planes or laptops, from parts with shapes, we do Not need to use the impractical time-consuming molecular dynamics, or quantum mechanics which just obstructs science.
So if we treat molecules or atoms as real objects (= parts ) with shapes (= discarding useless quantum mechanics, MD methods ), we can design and build any useful molecular devices by manipulating individual atoms or molecules (= real objects with shapes ) through atomic force microscopes with multiple probe tips, like building useful cars, planes and laptops from parts with shapes.
This current impractical time-consuming molecular dynamics based on shapeless quantum mechanical atoms is unable to simulate motions of atoms or molecules manipulated by the multi-probe atomic force microscopes as real objects with shapes, which is why atomic force microscopes have been deadend, useless with only one probe tip for more than 30 years
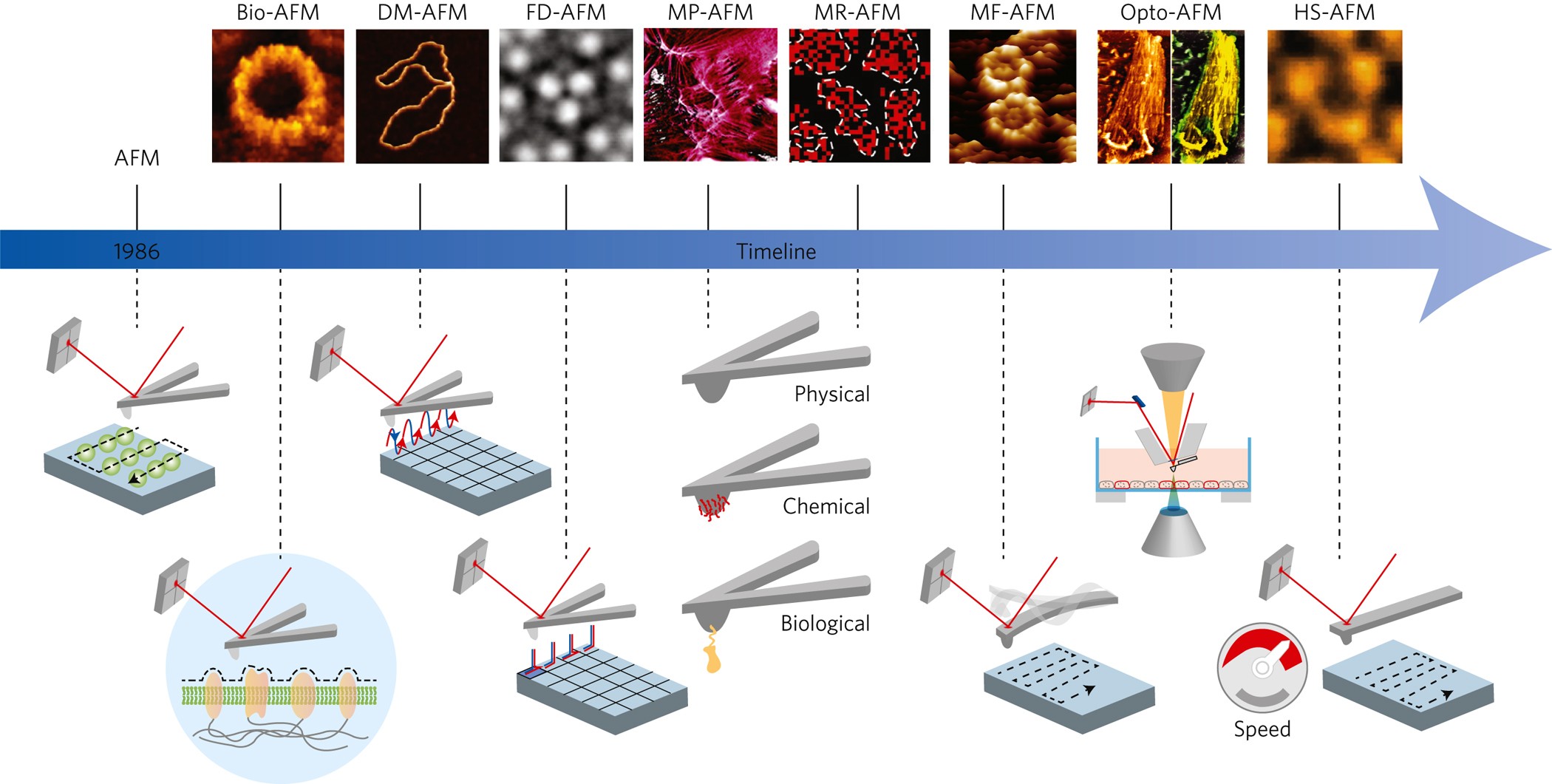
(Fig.4') Unphysical quantum mechanical shapeless atoms, impractical MD model has forced scientists to use the useless atomic force microscope with only one probe tip for 40 years. ← Our nano-technology is deadend, No progress from the useless single probe tip.
In all researches using today's atomic force (or scanning tunneling ) microscope with still only one (useless) single probe tip, scientists always try to express the atoms and molecules as the unphysical quantum mechanical shapeless atom, one-pseudo-electron DFT and the impractically-time-consuming molecular dynamical model (= MD, this-p.6-right-upper, this p.10-8. ) in vain.
↑ These current mainstream quantum mechanical DFT (= density functional theory ), and MD can give only useless pseudo-potentials or force field potentials instead of giving concrete shapes to atoms or molecules ( this p.15-16(or p.13-14), this p.34(or p.13 ) ).
This p.37(or p.16)-1st-paragraph says
"The timescale limitation of
MD simulation precludes reproducing this motion pattern. Instead, the tip moves
only forward and the depth is reported" ← The impractically-time-consuming MD model restricts the researchers' free manipulation of atoms.
Surprisingly, even the present atomic force (or scanning tunneling ) microscope still has only one useless probe tip, even after 36 years have passed since researchers could first manipulate single atoms by the microscope with only one tip in 1989. ← Our nano-technology is deadend, No progress from the useless single probe tip.
↑ So the unphysical quantum mechanics and molecular dynamical shapeless atomic model prevents the developing useful nano-technology by preventing developing (useful) atomic force microscopes with multiple probes.
↑ In other words, physicists (and academia ) fear that the useful multi-probe atomic force microscopes freely grabbing single molecules with shapes will make the present (useless) quantum mechanics and MD (= unphysical shapeless atomic model ) obsolete and unnecessary.
So physicists (and academia ) intentionally stop developing useful multi-probe atomic force microscopes that will eventually ditch the useless quantum mechanical or MD model, though they already have the technology. Today's atomic force microscope with only one probe tip is useless except for publishing papers in academic journals.
(Fig.5) Time-consuming molecular dynamics (= MD ) tends to rely on rough approximation = coarse-grained MD with pseudo-atoms sacrificing atomic-level accuracy.
Coarse-grained molecular dynamics (= CGMD ) approximation treating multiple atoms as one pseudo-atom or fictional beads (= This CGMD also has only pseudo-potential, no atomic shape, this p.2 ) is less time-consuming, but more inaccurate, sacrificing atomic-level resolution ( this-last, this p.1-right-last-paragraph, this p.23-5, this-introduction-3rd-4th-paragraph, this p.9-limitation ).
This p.2-1st-paragraph says
"Although coarse-grain simulation techniques can be used to somewhat avert
the time scale limitation, some of the fine (and perhaps critically important) details of atomic interactions
are lost during coarse-grain simulations"
This p.2-left-2nd-paragraph says
"the
coarse-grained models lack the atomic details of the interactions"
This p.2-introduction says
"However, owing to the loss of information, capturing atomistic properties through CGMD (= coarse-grained molecular dynamics ) or simply CG simulations is a challenge"
This p.1-intro-1st-paragraph says
"many CG (= coarse-grained ) methods suffer from an inability to correctly predict more than one or two
material properties"
Even this inaccurate approximate coarse-grained MD is also impractical, too time-consuming by moving pseudo-atoms little by little by differentiating pseudo-potentials artificially adjusted for CGMD ( this p.5-Fig.2 = simulation of just 1 μs CG molecular model took a day, this p.3-coase grain molecular dynamics ).
Accelerated MD is also too time-consuming to simulate milli-second molecular motion ( this p.20= just 30ns simulation, this p.6 = just 2500ns simulation, this p.15 = just 3.6μs simulation ). ← cannot be used for explaining actual long biological reactions.
Monte-Carlo (= MC ) method just tries to move individual atoms randomly, which often gives inaccurate results ( this p.2-right-last-paragraph ).
This Monte-Carlo tends to take much more time than the time-consuming molecular dynamics to simulate proteins, so useless and inefficient ( this p.2-right-1st-paragraph ).
This p.1-left-3rd-paragraph says
"A simulation of protein bovine
pancreatic trypsin inhibitor has shown that MD (= time-consuming molecular dynamics ) can be as
much as 10 times more efficient than MC (= Monte-Carlo moving atoms randomly is much more time-consuming, useless after all )"
Ab-initio (or first-principle ) molecular dynamics (= AIMD or FPMD ) including Car-Parrinello molecular dynamics (= CPMD ) based on unphysical quantum mechanical one-pseudo-electron DFT pseudo-potential (= DFT is fake ab-initio unable to predict anything ) is much more time-consuming and impractical than the traditional (= classical ) MD ( this p.2-I.introduction, this p.2-left-1st-paragraph, this p.23-middle ), so useless.
This p.19(or p.13)-3rd-paragraph says
"several cases are known DFT fails. the computational demands of CPMD (= ab-initio MD ) are very high.. limiting the length of the simulation to (only) 10-100 picoseconds (= ps, this p.5-right )
Exchange around polyvalant transition metal ions, takes place on a much longer timescale and is currently beyond the reach of CPMD (and of classical MD ).
"
Ab-initio MD or CPMD is useless, unrealistic, just artificially choosing (unphysical) one-pseudo-electron density (= DFT ) = unphysical plane wave electron's cloud whose coefficient parameters (= lacking real electron picture ) are artificially gradually changed at short time intervals ( this p.41,44 ) by using fictitious electron mass ( this p.20, this p.2-left-last-paragraph ), instead of moving real electrons (= only nuclei are moved classically, paradoxically, this p.1-left-3rd-paragraph ).
(Fig.6) Molecular dynamics (= MD ) with shapeless atoms with No boundaries can Not know when two atoms collide. → Updating each atomic position at short time intervals repeatedly (= to avoid two atoms clashing and overlapping ) takes too much time.
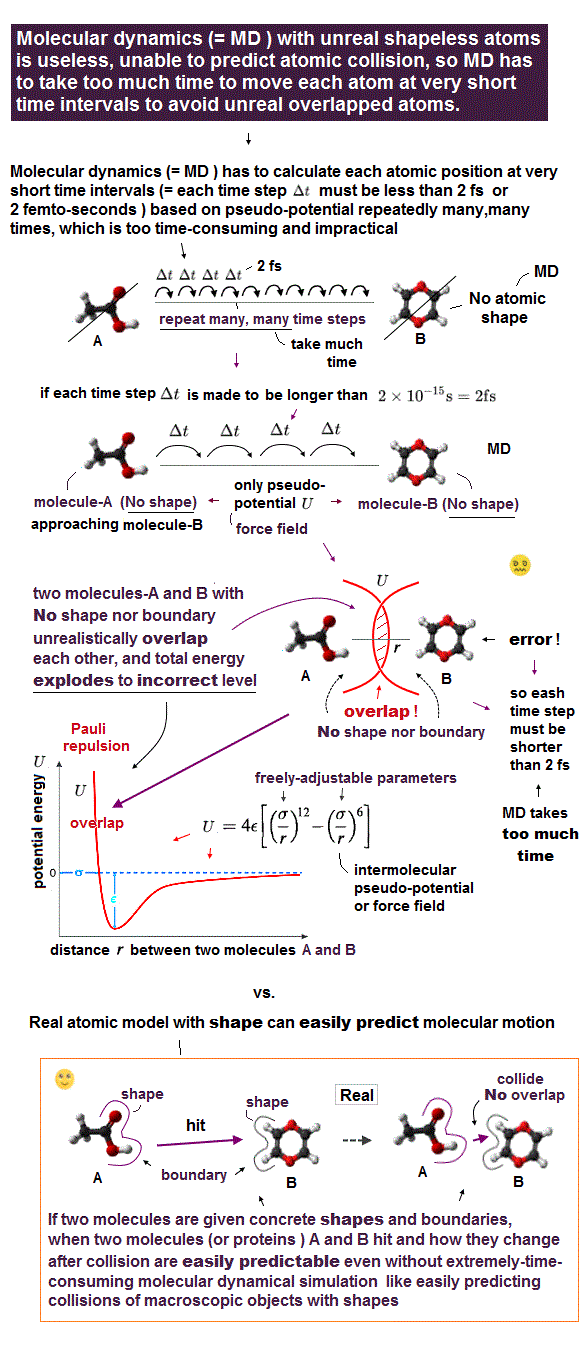
Today's only method of simulating motions of molecules and proteins is the impractical molecular dynamics (= MD ) using only artificial pseudo-potential called force fields with No atomic shapes.
In real objects with shapes, we can easily predict the objects' motion and when two objects with shapes collide, like predicting billiard balls' trajectory.
But in the unrealistic shapeless quantum mechanical atoms and MD, the time when two shapeless atoms collide with each other is unpredictable due to lack of individual atomic shape and boundary.
So in this impractical molecular dynamical simulation (= MD ), researchers have to calculate and move each atomic position little by little at extremely short time intervals (= each time step Δt must be less than only 2 fs = 2 femto-seconds = 2 × 10-15 seconds, this p.4-2nd-paragraph ) repeatedly many many times ( this or this p.5-p.29 ).
↑ Just simulating 1μs molecular motion must repeat calculations of atomic positions as many as 109 times (= 1 μs/ 1 fs = 1 × 10-6 s/ 1 × 10-15 s ), which unrealistically takes too much time, and hampers science.
If they choose each time step longer than 2 fs, it increases the chance of two shapeless quantum mechanical atoms colliding and unrealistically overlapping each other.
↑ Because during longer time step, all atoms can move longer distance at once, which increases the risk of unfavorable atomic collision or overlap (= of two atoms ) causing errors and wrong results called "energy explosion or blow up ( this p.37-1st-paragraph, this p.27-28, this 5th-paragraph, this p.28-1st-paragraph, )".
This p.10-2~3rd-paragraphs say
"Too large a time step can cause a molecular dynamics simulation to become unstable, with the
total energy rapidly increasing with time. This behavior is often colloquially termed "exploding"
and it is caused by devastating atomic collisions that occur when a large time step propagates
the positions of two atoms to be nearly overlapping; the (Pauli) repulsive interactions then create a
strong force that propels these atoms apart"
This or this-p.1-lower ~ p.2 says
"if a very small value of time step is used, it will not be
efficient since it takes a very long calculation time. However, unfortunately, the MD integration algorithm becomes
unstable at large time step size. If too large a time step τ is used, the motion of particles becomes unstable due to the very big truncation error in the integration process, so the total energy of the system may rapidly increase with time.... This behavior is often called exploding and caused
by devastating atomic collisions that occur when a large time step.. nearly
overlapping"
↑ As a result, the MD simulation based on unphysical quantum mechanical shapeless atoms has to choose very short time step (< 2fs ), and repeat those very short time steps, many, many times to avoid erroneous overlap of MD's shapeless molecules, which takes impractically much time, hindering scientific progress.
The 2nd, 6th, 8th paragraphs of this recent news (7/9/2025) say
"The team's calculations reveal that the potential for errors caused by using a 2 (or more) femtosecond (= 2fs ) time step is even greater than they had anticipated."
"using anything greater than a 0.5 femtosecond time step can lead to violations of equipartition... can introduce error"
"using a smaller time step inevitably increases the computational cost."
↑ This recent research found today's MD must take much more time by choosing smaller time step of 0.5fs instead of 2.0fs to avoid errors, which is much more time-consuming and impractical.
The 7th-paragraph of this hyped news (7/9/2025) says
"You usually need to use some tricks to get the molecular dynamics simulations (= time-consuming, impractical ) to crystallize. We were even more amazed to observe graphite crystallizing spontaneously at pressures up to 15 GPa—conditions where diamond should be the stable form"
↑ This research paper ↓
p.2-left-last~right says "Here we compute the graphite-liquid and diamond-liquid coexistence lines,.. MD (= molecular dynamics ) simulations using.. (neuroevolution potential, NEP) and trained on energies and forces computed at the level of density functional theory (DFT) either in the local density approximation (LDA) or with a generalised gradient approximation (GGA) functional with non-local van der Waals correlation ‘OptB88-vdW (= whose parameters were fitted to experiments, this-p.3-left )"
p.3-left-3rd~4th-paragraphs says "with NEP@LDA (= artificially-created MD potential based on DFT-LDA exchange functional ) overestimating and NEP@OptB88-vdW underestimating the density compared to experiments" ← simulation results of quantum mechanical DFT and MD were wrong.
"MD simulations customarily overestimate the stability of liquid phases"
p.4-right-last-paragraph says only 20ns simulation of graphite or diamond was conducted due to the extremely time-consuming MD, which is useless.
↑ The force field potential of the impractical MD was trained on the unphysical DFT where differently-chosen exchange-correlation potential functionals such as LDA, OptB88-vdW.. gave different erroneous results.
This-p.2-left-2nd-paragraph says
"the
accuracy of pseudopotential based calculations does depend
on the quality of the pseudopotential itself. Simulations
of systems in high-pressure conditions have to be
checked carefully in this respect since interatomic distances
can get much shorter than at ambient conditions;
hence it is often necessary to reflect this in the pseudopotential
design (= artificial )"
↑ So quantum mechanical DFT and MD results depend on artificially-chosen exchange-correlation potential functionals (= LDA.. ), different pseudo-potentials (= under different pressures ), wavefunctions, which can Not predict nor clarify any physical mechanisms such as graphite, contrary to hypes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}