
Home page
AI bad artificial enzymes
AI cannot cure cancers
(Fig.A) Artificial intelligence (= AI ) is hyped, useless for drug development
Contrary to an incredible amount of overhyped fake news, it is impossible to use artificial intelligence (= AI ) for developing effective drugs or treatments that do Not exist in its present (old) training data sets.
The 1st, 7th, 9th paragraphs of this site says
"More and more people are starting to decry the lack of progress from AI-driven companies"
"Whatever experimental data the traditional pharma companies made public was the dataset AI-driven companies inherited. But this dataset is problematic"
"Remember, AI will only produce similar compounds to the examples on which it was trained (= developing new effective cancer treatments is impossible )"
This-3rd-paragraph (9/10/2025) says
"yet there are few AI-discovered candidates in late-stage clinical trials, and not one has been approved ( this-abstract )"
The 1st, 7th paragarphs of this site (6/25/2025) say
"AI won't revolutionize drug discovery for Big Pharma. They don't need it."
"Breathless headlines promise that AI will slash drug development timelines.. these claims consistently fall flat. Even if we could design perfect binders instantly (which we can't do - yet), that barely move the needle."
The 3rd, 9th paragraphs of this news (1/3/2025) say
"AI-generated drugs have yet to demonstrate an ability to address the 90% failure rate of new drugs in clinical trials.... its effect on drug development remains unclear"
"the AI in drug development is constrained by small, low-quality datasets. It is difficult to generate drug-related datasets on cells, animals or humans for millions to billions of compounds. While AlphaFold is a breakthrough in predicting protein structures (= only static protein, unable to predict molecular, protein conformational change nor drug effectiveness ), how precise it can be for drug design remains uncertain."
The 6th paragraph of this site (1/16/2025) says
"AI methods are still going to be subject to the same white-knuckle dice-rolling as all the others when they get into human trials, because we have (as yet) No computational tools that really help us predict whether we have picked the right target, the right disease, the right biochemical pathway, or the right compound to affect it without doing anything unexpected along the way. "
↑ Overhyped AI can Not predict any drugs at all.
This 3rd, 4th, 9th paragraphs say
"And AI-first methods can only be trained on that existing experimental data. It is like exploring a drop of water in an ocean, he said."
"In addition, even within that limited available experimental data, a sizable portion is of questionable quality and often not reproducible"
"It is virtually impossible for current AI approaches to find breakthrough novel drugs unaided, he explained. And when you look at the companies who purportedly 'discovered' drugs with AI, you find that most have developed small 'me-too' modifications of well-trodden drug structures."
This 2nd-paragraph says
"While biotechnology companies have been touting a decade-old narrative about AI drug discovery being exponentially faster and cheaper than conventional drug discovery (= hype ),.. none have made it through Phase III and FDA approval yet" ← "AI drug discovery" turned out to be overhyped illusion.
This (or this or this ) 4th paragraphs say
"AI has really let us all down in the last decade when it comes to drug discovery,.. We've just seen failure after failure"
This 3rd-paragraph says
"(AI) algorithms and systems of care have yet to improve patient outcomes at scale" ← AI has No benefit for medical treatment.
This 5th-paragraph says
"There is Not yet an approved drug originating from AI tools" ← AI has contributed nothing to drug discovery.
This-What are the implications?-2th-paragraph says
"AI is far from being able to assist in the entire process of drug creation or remove people entirely from it. There are still questions over its reliability, too,"
This p.30-5 Challenges and future perspective-2nd-paragraph says
"yet they (= AI ) are still far from having human intuition... it cannot replace humans. AI models are not perfect and can have detrimental
limitations, such as false positive or false negative predictions, especially when dealing
with unfamiliar cases... AI is highly dependent on the quality of the training data."
This 12-13th paragraphs say
"it's still early days...
Generative A.I. is transforming the field, but the drug-development process is messy and very human."
This 9th-paragraph says
" AI has great potential (= still unrealized ) to revolutionize drug discovery, it still faces significant hurdles in accurately predicting drug efficacy."
AI, Alphafold trained on the static protein structure database (= PDB ) obtained by traditional old methods (= X-ray crystallography.. ) can Not predict proteins' dynamical behavior or true biological functions. → No drug discovery.
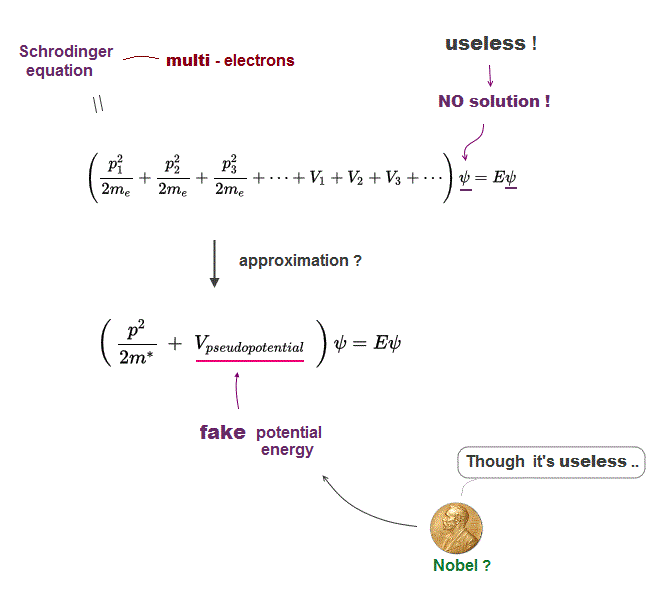
Because the unrealistic quantum mechanical shapeless atomic model prevents researchers from designing useful molecular nano-machines by using atomic force microscopes with multiple probes.
↑ The current deadend quantum mechanics tries to prevent researchers from developing useful multi-probe atomic force microscopes, though we already have the technology of manipulating individual atoms and clarifying protein behavior at atomic level, if we want to do it.
(Fig.D) Success rates of designing de novo proteins bound to known target peptides are too bad (= 6 % ~ 50 % ) to use for drug development.
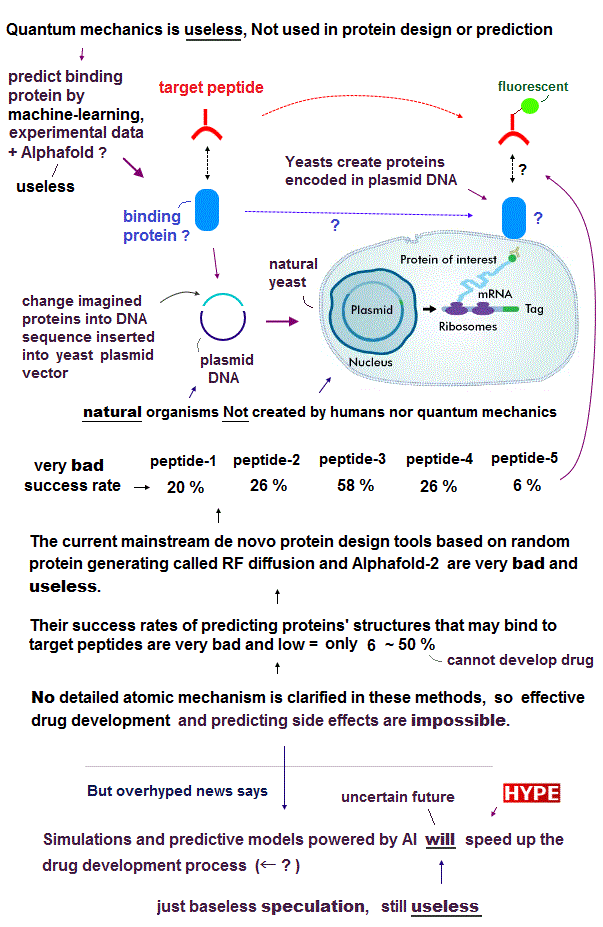
According to the latest (exaggerated AI) research published in Nature, the current mainstream (useless) de novo protein design method just tried to randomly generate noise or new amino acids against some known target peptides (which method is called generative AI or RF diffusion, this 7th-paragraph = noise, this p.5-Peptide binder design ) combined with the Alphafold-2 (= AF2 ) predicting protein structure, and other alleged machine-learning methods (= ProteinMPNN, Hallucination, RFjoint inpainting ). ← No (useless) quantum mechanics is used.
These so-called AI and machine-learning methods such as Alphafold (= AF ) and RF-diffusion are trained on the already-known (static) protein structure dataset (= Not on quantum mechanics, this 4~5th-paragraphs ) without looking into individual atomic interaction nor dynamical protein conformational change.
↑ Even RF-diffusion randomly generating amino acids and ProteinMPNN finding protein sequences from the protein structures rely on experimentally-obtained protein structure databank (= PDB, this-p.68(or p.67)-4.1, this-p.3-protein-MPNN ) without considering atomic mechanism nor relying on the useless quantum mechanics.
↑ It is impossible that today's overhyped AI, machine-learning or Alphafold without looking into real atomic interaction predict actual protein behavior or discover effective drugs.
Actually even in this latest AI methods such as RF diffusion, their success rates of predicting de novo proteins binding to target peptides were very bad and low = only less than 20% ( this-abstract, this-lower-Capabilities-3rd-paragraph ).
This p.6-Discussion-3rd-paragraph says
"there is considerable
room for improvements,... and the success rates are still quite low"
In this latest AI de novo protein research, they inserted DNAs allegedly encoding these new proteins (= expected to bind to some target molecules ) designed by the random RF-diffusion and Alphafold-2 into yeast plasmid DNA vector, and get (natural) yeasts to generate those artificially-designed proteins on their cells' surface.
If their protein design or prediction was right, those newly-produced proteins could bind to some chosen target molecules or peptides labeled with fluorescent (= biotinylated ) substances in screening using yeast displaying proteins ( this p.2(p.1), this p.5-left-2nd-paragraph ). ← Precise atomic interactions between proteins or where exactly of these new proteins bind to peptides are unknown, though.
In these methods, they designed (= predicted ) and produced 96 new different binder proteins for each (natural known) target (molecule) peptide such as neuropeptide Y (= NPY ), glucagon (= GCG ), parathyroid hormone (= PTH ), and apoptosis peptide called Bim ( this 4th-paragraph ).
Success rates of predicting proteins bound to target peptides are very bad = only 20% (= 20/96 ) in NPY, 26% (= 25/96 ) in GCG, 58% (= 56/96 ) in PTH, 26% (= 25/96 ) in Bim.
See this-p.5-left-2nd-paragraph, p.5-left-last~p.5-right-1st-paragraph or this p.6-1st,3rd-paragraphs,
↑ This p.5-left-2nd-paragraph says
"Initial
screening with yeast surface display revealed quite high (= Not so high ) binding success
rates, with 25 out of 96 designs binding GCG (= success rate was very low = only 25/96 = 26% ), and 20 out of 96 binding
NPY (= success rate was very low = only 20/96 = 20% ) at 10 nM peptide concentration"
In the case of designing protein binders to secretin (= SCT ) peptide, its success rate is worst, only 6 % (= only 4 proteins bound to the target SCT peptides out of total 66 proteins designed to bind to SCT peptide, this p.4-1st-paragraph )
↑ This p.3-left-3rd-paragraph says
"Initial screening using yeast surface display identified 4/66
binders for SCT (= success rate was very low = only 4/66 = 6% )"
↑ Only 6% ~ 20% successful protein prediction rate, which cannot be used for effective drug development or predicting exact side effects.
↑ To precisely predict side effects, interactions with many other peptides, proteins, substances inside bodies must be precisely predicted, which is impossible in this very bad-success-rate RF-diffusion + Alphafold-2.
The 9th, 2nd-last paragraph of this hyped news (7/1/2025) say
"First, they ask the AI tool how to improve a desired enzyme's performance. The AI tool searches datasets of known enzyme structures (= from experiments ) and suggests sequence changes (= this AI's prediction based on known enzymatic datasets is bad )....."
"..The automated protein-building machines at the iBioFoundry produce the suggested enzymes, which are then rapidly (experimentally) tested to characterize their functions. The data from those tests are fed into another AI model (= so experimental tests of enzymatic functions were needed after all. The overhyped AI alone can Not predict anything ), which uses the information to improve the next round of suggested protein designs."
"Next, the researchers plan to (= still unrealized ) continue improving their AI models and upgrade equipment"
↑ This-research paper's p.3-right-2nd-paragraph says
"By combining the two unsupervised models ( trained on experimentally-known datasets ), we generated a list of
180 variants each of AtHMT and YmPhytase for initial screening. We found that 59.6% of AtHMT and 55% of YmPhytase
variants performed above the wild type baseline" ← So even the latest AI could improve the original wild enzymatic function by only 5.0% (= 55% ) ~ 9.6% (= 59.6% ), which success prediction rate was too bad and useless.
p.4-Fig.2 and p.8-left says
"For each round of engineering, we trained a supervised prediction
model based on all experimentally measured variant fitness data from
current round and all rounds prior"
↑ So they just improved the enzymatic functions by actually observing experiments (= mutants functional assay ), Not by AI.
p.7~p.11-methods shows this research used only macroscopic biological methods without manipulating single molecules nor using (useless) quantum mechanics.
So the overhyped AI is far from improving actual enzymatic functions.
The 3rd, 7th--paragraphs of this hyped news (7/8/2025)
say
"broadening capabilities to screen potential new (= still unrealized ) materials with machine learning models"
"Density functional theory and the faster machine learning models trained on it, for example, can compute energy, forces, and thermodynamics. They lack spin degrees of freedom. Without that additional set of variables needed to predict magnetic properties, existing methods are inaccurate, too slow, or too expensive for designing magnetic materials." ← The existing quantum mechanics failed to predict spin magnetic moments of material.
↑ This research paper ↓
p.7-Discussion-2nd-paragraph says "We found a data quality issue in the MP dataset caused by suboptimal DFT computational settings, which led to non-converged magnetic moments and subsequently inconsistent labels"
↑ They trained it on bad datasets, so this hyped machine-learning or AI was useless, unable to predict anything after all.
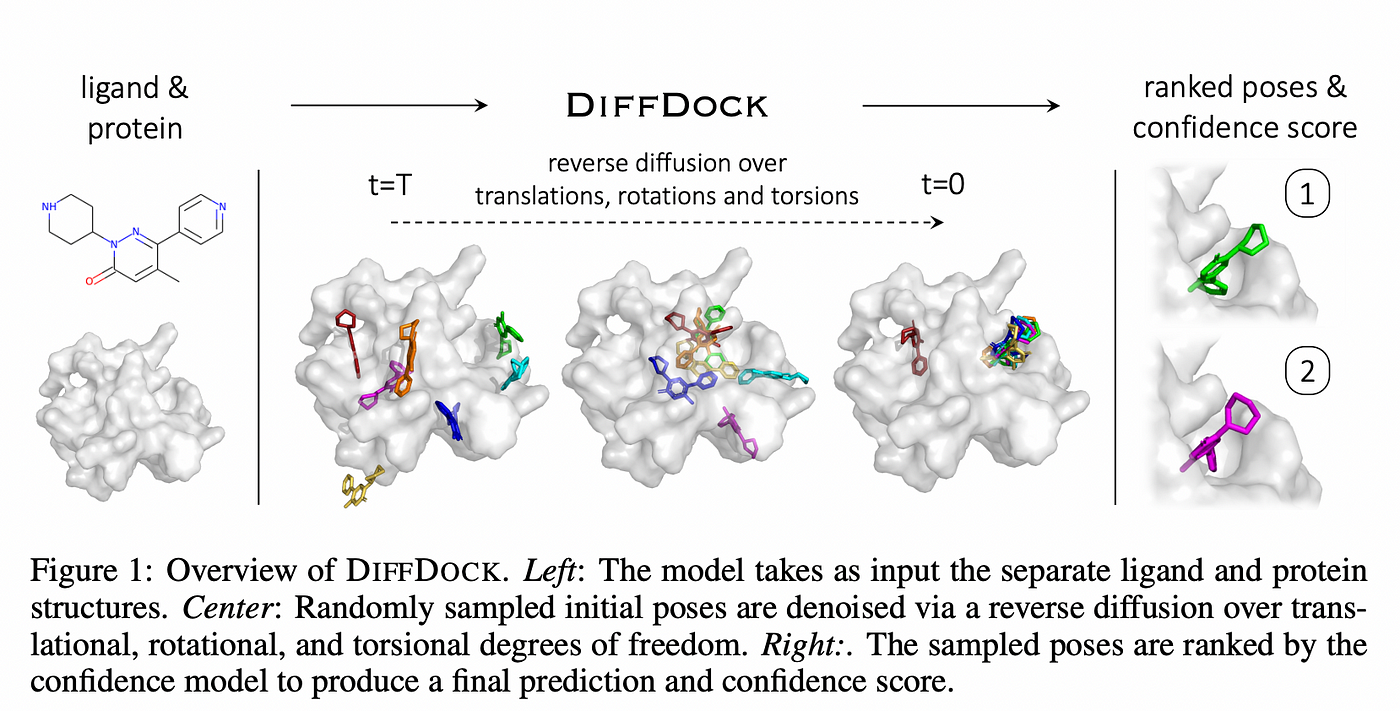
Microsoft's ad-hoc protein diffusion model (= RF diffusion, Evodiff, Diffdock.. ) also just tries to design and generate somewhat (meaningless) random (noise) proteins' sequences and structures against some target molecule (= static useless protein like Alphafold ) by (overhyped dubious) AI (= Not quantum mechanical prediction, this 7th-paragraph ).
↑ This protein binding prediction rate is still impractically low (= only less than 19% even with the help of Alphafold-2, this p.8-Design of protein biding, p.9-lower, p.10-Fig.6b, this 9th-last paragraph, this p.3-last-paragraph ), and lacking many experimental verification ( this 8,13th paragraphs, this-lower Conclusion, this 7,11th-paragraphs, this p.1,p.2-right ), so it's also useless for drug development contrary to the media-hype.
↑ These are typical examples preventing real scientific development or curing diseases by overhyped AI that just wastes huge taxpayers' money and time only in these fruitless science (= whose only purpose is to publish papers in top journals instead of curing diseases ) which just makes people wrongly believe the present science may be progressing (= actually Not ).
(Fig.B) AI success rate of predicting proteins binding to target bilin molecule was very bad = only 9.6%
The 1st, 4th, 5th, last paragraphs of this hyped news say
"group at the University of Washington, have developed a novel deep learning method, RoseTTAFold All-Atom (RFAA), for prediction and design of complexes of proteins, small molecules, and nucleic acids. Subsequently, they developed RFdiffusionAA, which builds protein structures around small molecules (= still No ability to predict proteins accurately, though )."
"It was already known that bilins are optically featureless unless they are held within a defined binding site, at which point they become intensely colored and emissive. In this work current Ph.D. student.. devised a multiwell assay that could screen many RFdiffusionAA-generated genes in parallel, using E. coli cells that could make phycoerythrobilin (PEB)."
"Morey-Burrows evaluated 94 designs in one go, with the multiwell assay revealing visibly colored cells in nine wells (= only 9 proteins out of 94 proteins designed by deep learning seemed to bind to the target bilin, very bad prediction rate and useless for drug development ). He had identified nine proteins dissimilar to each other and to any native bilin binder, based on pigmentation or fluorescence."
"fluorescent reporter probes with tunable excitation/emission maxima would (= uncertain future, still useless ) be useful biochemical tools."
↑ This research tried to predict proteins binding to target molecules (= such as bilins ) by using the deep learning ( or AI ?) tool called RoseTTAFold All-Atom (RFAA) trained on experimentally-obtained structure data such as PDB (= useless quantum mechanics was Not used ), and its successful prediction rate was only 9.6% (= only 9 proteins out of 94 designed proteins barely bound to the target bilins ), which low success rate cannot be used for actual drug design that needs precise prediction of many proteins' interactions to avoid serious side effects.
↑ This research paper ( this ↓ )
p.1-Abstract says "AlphaFold2 (AF2) and RoseTTAFold (RF).. they are unable to model covalent modifications or interactions with small molecules and other non-protein molecules that can play key roles in biological function"
p.4-Training RFAA says "From the PDB, we curated a protein-biomolecule dataset including protein-small molecule.. we supplemented the training set with small molecule crystal structures from the Cambridge Structural Database" ← They trained or did "deep learning" based on experimentally-obtained protein, molecule database, Not on (useless) quantum mechanics.
p.10-2nd paragraph says "Starting from a panel of 94 RFdiffusionAA designs and using phycoerythrobilin (PEB) as the chromophore, we identified nine bilin-binders based on visible pigmentation or fluorescence in a whole cell screen (a 9.6% hit rate )" ← This deep-learning successful rates of predicting proteins binding to the target bilin molecules is very bad, only 9.6% = 9/94.
↑ To develop or find effective drugs, we have to precisely predict interactions between drug molecules and many proteins inside bodies to avoid the serious side effect, so this extremely-low prediction rate of protein-molecule interaction (= only 9.6% ) can Not be used for practical drug design.
The point is these deep-learning or AI methods do Not consider the detailed atomic or molecular interactions, instead, they just tried to train AI tools based only on the experimentally-observed (final static) protein or molecule structures (= without considering the "process" of how each protein was folded through atomic interaction due to the current mainstream physics unable to give realistic "shapes" to atoms ).
This is why the current biological and medical researchers are still unable to utilize practical atomic interactions for drug development, and they just waste their time only to publish papers in academic journals whose useless pseudo-science ruins their careers and future.
(Fig.A) AI's success (= hit ) rate of predicting molecules binding to target proteins was very bad, only 14%, which cannot be used for drug development

The 2nd and last paragraphs of this hyped news say
"they can screen multi-billion compound libraries against two unrelated targets (= only two targets, which cannot be generalized ), a ubiquitin ligase target KLHDC2 and the human voltage-gated sodium channel NaV1.7."
"The researchers believe that the future enhancements to their protocol will (= just speculation, still useless research ) likely involve the integration of GPU acceleration and deep learning models."
↑ This research paper ↓
p.1-abstract says "we discover hit compounds, including seven hits ( 14% hit rate ) to KLHDC2 and four hits ( 44% hit rate ) to NaV1.7" ← Success (= hit ) rates of predicting molecules binding to target proteins by this AI were very bad = only 14% and 44%
p.4-right-3rd-paragraph says "Finally, ten molecules with Tanimoto similarities of less than 0.33 to the known inhibitors of NaV1.7 from the ChEMBL database43,44 were selected for synthesis. Of these, nine were successfully synthesized, and their activities were measured using the whole-cell patch-clamp electrophysiology assay on hNaV1.7 channel stably expressed in HEK-293 cells... IC50 values of better than 10 μM were observed for four compounds, translating to a hit rate of 44.4% (= 4/9 )"
↑ This p.3-Reviewer #2's 2nd-paragraph says "but I tend to agree with their argument that it is unknown how well these model generalize outside the benchmark sets. "
↑ This method of screening molecules bound to target proteins was applied to only two proteins with low success rate (= only 14% and 44% ), which cannot be used for drug discovery.
The 6th, 8th, 11th paragraphs of this hyped news say
"the method has limitations: It allows scientists to model proteins only in a static state at a specific point in time." ← Alphafold cannot simulate molecular motion nor conformational change.
"In order to match protein targets to drugs to treat cancer and other diseases, we need a more accurate understanding of these physiological changes" ← this change cannot be explained by Alphafold that just gives static protein structures.
"In this study, the researchers were able to manipulate the evolutionary signals from the protein to use AlphaFold 2 to rapidly predict multiple protein conformations, as well as how often those structures are populated." ← This research artificially manipulated parameters of Alphafold2 to change some proteins' conformation.
↑ This research paper ↓
p.4-left-1st-paragraph says "none of the Src predictions were found to be in the I2 state, although the enzyme is known to infrequently occupy this conformation. This suggests a resolution limitation in using AF2 (= Alphafold2 prediction was wrong )"
p.5-left-1st-paragraph says "First, the M290L predictions (= by Alphafold2 ) are inaccurate. Specifically, the effects of the mutation on the ground state population are predicted to be the opposite of those seen in experiments"
p.8-left-2nd-paragraph says ", our method did not achieve perfect accuracy. Specifically, it failed to predict the significant changes in GMCSF’s N-terminus dynamics associated with the H83N mutation. Additionally, our method failed to replicate the significant changes in dynamics of residues 80–90 for the H83Y mutations" ← many failures in Alphafold-2's prediction
p.10-right-4th-paragraph says "our capacity to predict relative state populations without prior knowledge was limited for a few protein systems within the test set" ← This method was applied to only a few proteins.
↑ This p.3-[1] and [2] (= reviewer's comment ) say "The abstract is a bit too positive given that it makes no mention of the issues that the method encountered"
"How can one be sure that the method presented in the paper is not overfitted to Abl1 kinase or to the granulocyte-macrophage colony stimulating factor ? How should one prove that the method is generalizable to all proteins ?" ← This research artificially manipulated parameters of Alphafold-2 for only a few proteins, which cannot be generalized nor applied to other proteins.
(Fig.T) AI or Alphafold2 success rate of predicting transmembrane proteins was very bad = only 29%
The 2nd, 4-5th, last paragraphs of this hyped news say
"However, current approaches to engineering nanopore sensors are limited to naturally occurring proteins (= just using natural proteins, cannot design them ), which have evolved for very different functions and are less than ideal starting points for sensor development."
"With the help of computational design, the researchers developed methods to design stable nanopore channels with tunable pore shapes, sizes, and conductance (= this research's computational method had a bad success rate of designing target proteins )"
" Now we have shown that we can successfully design nanopores with a high success rate (= No. this research had low success rate, so useless ), which have stable and reproducible conductance"
"Although we are still quite a bit removed from this point (= still useless ), the researchers envision a future (= just speculation ) in which portable devices with different nanopores can sense a range of metabolites, "
↑ This research paper ( this ↓ )
p.6-middle says "All 16 such designs failed to express in E. coli (= 16 proteins designed or predicted by this method failed to be created when their DNA sequences were encoded into plasmid DNA expressing proteins inside E.coli )"
p.7-14th~ sentences say "Therefore, we selected 4-10 designs per blueprint for which AlphaFold2 (= based on experimentally-obtained PDB protein database, Not on quantum mechanical prediction ) predicted high-confidence structures closely matching the design models"
p.9-6th-sentence~ says "The 12-strand TMB12_3 inserted successfully into the membrane, producing distinct jumps of current of reproducible intensities, and had stable nanopore conductance. While the design TMB10_163 did Not have detectable nanopore activity (= this method failed to predict nanopore protein structure )"
p.12-8th-sentence says "Based on the intensities of the current jumps, we estimated the conductances of single-channel events, which increased with pore size (= if nanopore was successfully formed, electric conductance was measured )"
p.31-upper mentions "Custom genes in a pET29b (= plasmid DNA ) vector (into which the designed protein's gene was encoded to express the protein in E.coli )"
p.34-last-paragraph mentions "Rosetta full-atom model energy function" which was parameterized based on experimentally-obtained molecular or protein energy data ( this-abstract ), Not on (useless) quantum mechanics.
p.36 also used Alphafold2.
p.39~p.50 of this shows this research designed the total 48 proteins, and 14 proteins could Not be produced (= fail, No expression ), 17 proteins were successfully expressed, but they did Not show pore activity (= 17 designed proteins failed to show electric conductance ).
Only 14 designed proteins barely showed pore activity (= conductance ).
So the total success rate of producing the designed proteins was very bad = only 14/48 = 29%, which low success rate can Not be used for effective drug design (= their proteins' precise properties and toxicity were still unknown and unpredictable ).
The 4th, 9th paragraphs of this hyped news (7/9/2025)
say
"By using AI in this way,.. paving the way for faster, more affordable drug development and diagnostics that could (= just speculation, still unrealized ) transform biomedical research and patient care."
"we can engineer proteins to bind a specific target site or ligand, as inhibitors, agonists or antagonists," ← hype
↑ This research paper ↓
p.1-abstract says nothing about drug development or diagnostics, contrary to the above hyped news.
p.7-left-1st-paragraph says "Using an AlphaFold2 model of ChuA (= E,coli's membrane protein ) as a target, we utilised RFdiffusion and ProteinMPNN to design binders targeting.. ChuA,... ( this-p.2 )
".. We generated ~20,000 ChuA binders in silico and selected 96 designs for wet lab screening using AlphaFold2 filtering and manual curation (← AI alone cannot predict anything )... We screened these binders for their ability to inhibit the growth of E. coli.... with eight binders displaying more prominent zones of inhibition"
p.8-Fig.5a shows only 8 binders (= A10, C8, D8, G7.. ) out of 96 selected binders predicted by AI bound to the target ChuA protein. ← extremely low success rate (= 8/96 ) of AI (= still their drug effectiveness is unclear ).
p.9~ Methods used only macroscopic biological tools without manipulating single atoms nor relying on useless quantum mechanics.
All these AI tools such as RF diffusion randomly generating binder's backbone, ProteinMPNN predicting protein's sequence from the protein structure and Alphafold are based on experimentally-obtained static protein databank (= this-p.68(or p.67)-4.1, this-3rd~5th-paragraphs, this-p.3-protein-MPNN ) Not relying on the useless quantum mechanics.

Feel free to link to this site.
{kind=link}