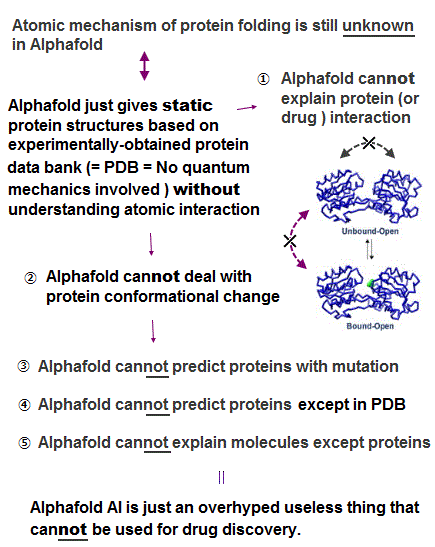
(Fig.1) AI-Alphafold is useless, unable to clarify any biological reactions.
Useless quantum mechanics cannot solve its Schrödinger equation nor predict energies of any multi-electron atoms or molecules, so all the current methods including AI, Alphafold cannot predict nor develop effective drugs, contrary to a lot of fake news.
The overhyped artificial intelligence (= AI ) is useless for discovering new effective drugs that are Not included in its training datasets based on experimental results ( this-12-13th-paragraphs ).
Alphafold (and other AI methods ), which is trained on the experimentally-obtained static protein structure data bank (= PDB = often wrong protein structures ), cannot predict new ( mutated, disordered ) proteins ( this-p.1-right-last-paragraph ) nor protein conformational change ( this-2nd-paragraph, this-31~32th-paragraphs, this-p.1-abstract ).
This or this-p.2-right-4th-paragraph says --
AI cannot predict mutation
"AlphaFold has some limitations. It is Not
designed to predict the effect of mutations,
such as those that cause disease, on a protein's shape. Nor was it trained to determine
how proteins change shape in the presence of
other interacting proteins, or molecules such
as drugs" ← Alphafold cannot discover drugs.
The 7-8th, 10th paragraphs of this (2025) say -- Alphafold useless
"AI oversimplifies the representation of the protein's flexible regions."
"AlphaFold is trained on static representations of protein structures, but many proteins, including AGP, are anything but static"
"while AI tools like AlphaFold are powerful, their training data lacks information about complex protein behavior, and results must be validated against experimental data"
This-p.6-Discussion says -- No experimental data
"AF2 (= alphafold-2 ) currently relies heavily on (X-ray structures in)
the PDB, as the quality of predicted structures dropped dramatically when we considered proteins for which no
template structure could be found"
This-1st~2nd-paragraphs (2025) say -- Unreliable experimental data
"The ground truth data that AlphaFold relies on comes from those very experiments. X-ray crystallography and cryo-electron microscopy (cryo-EM" ← These current experiments
cannot get
exact structures at atomic level ( this-p.9-3rd-paragraph ), so AI, Alphafold are useless forever.
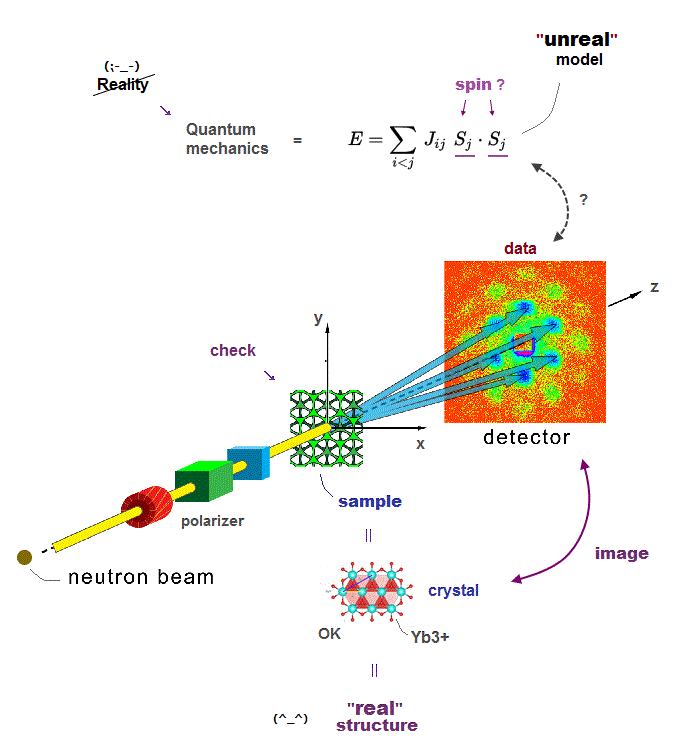
Only multi-probe atomic force microscopes can directly see and clarify atomic structures of proteins, which is hampered by today's unreal quantum mechanical shapeless atomic model.
This-p.14-conclusion says
"Predicting pathogenic variants in disordered regions
remains challenging"
This-lower-Limitation influencing Alphafold's capability (2025) says
"Furthermore, intrinsically disordered regions (IDRs) that lack stability in the structure are predicted with low confidence "
"The main difficulties associated with accurately predicting three-dimensional protein structures depend on the availability of training data,"
Alphafold's inability to explain molecular or protein conformational change means Alphafold cannot clarify any biological reactions ( this-p.1-abstract ), protein docking ( this-5-6th-paragraphs ), nor discover effective drugs.
The 4th-paragraph of this ( or this ) says -- Media hype
"That's why press coverage about how this new prediction database will revolutionise drug discovery are overblown. Protein structures shift and slide in the presence of small-molecule ligands, sometimes subtly and sometimes dramatically, but AlphaFold is Not (yet) equipped to predict these changes."
This-9th-paragraph (in 2025 ) says -- No drug discovery
"how precise it (= Alphafold ) can be for drug design remains uncertain." ← still Alphafold contributed nothing to drug discovery.
This-8th-paragraph says -- No drug prediction
"Knowing a protein's structure is a first step. But discovering new drugs requires understanding how that protein will bind to a novel drug structure. But there is No binding data for truly novel compounds, And tools like AlphaFold cannot make these predictions."
This-3rd-last-paragraph says -- No drug in alphafold
"We don't know yet whether drugs designed using AlphaFold will pass successfully through clinical trials, and none have been tested in humans yet"
Alphafold also cannot predict post-translational modification (= phosphorylation, sugar.. ) that is important for biological reactions ( this-lower-What Alphafold2 can't do, this-4th-paragraph ).
This-p.13-right-top says (2025)
"their inability to capture
rare or biologically significant phosphorylation-induced transitions necessitates
cautious interpretation. "
This-lower-Road ahead (2025) says
"Predicting protein interactions, understanding dynamics, and modeling the full complexity of cellular environments remain open challenges"
This or this-middle-Five years on-3rd-paragraph (2025) says
"But it's far from perfect... But AlphaFold is known to be less accurate at making predictions about multiple proteins or their interaction over time."
This-lower Known limitations and common fixes say -- No motion
"( The useless Alphafold-4 can predict ) One Structure Only: No flexibility or multiple conformations
- No Ligands or Cofactors: Cannot model metal, DNA, etc.
- Limited on Membrane Proteins: Sometimes topology is wrong"
This-lower-5.2-5th-paragraph says -- Useless Alphafold-4
"AlphaFold 4 moves toward ensemble and dynamic predictions, but limitations remain, as discussed in the literature: Nature article on AlphaFold 3" ← so Alphafold4 has the same limitation as the previous Alphafold-3.
"Approaches to address dynamics: Combine AlphaFold 4 outputs with molecular dynamics (MD)" ← The impractical time-consuming MD is the current only method of simulating proteins' motion.
(Fig.2) Alphafold-3 cannot predict protein or enzymatic conformational change.
Like all other AI, Alphafold-2, Alphafold-3 was also trained on the experimentally-obtained protein structure dataset of PDB, so cannot predict new unknown proteins or mutated cancer proteins, hence useless.
This site (2025) ↓
4th-paragraph says -- No protein motion
"AF3 (= Alphafold-3 ) produces a single, static conformation and doesn't model dynamics. Biology is dynamic; proteins flex and adopt different states that can affect drug binding. AF3 cannot simulate these motions."
5th-paragraph says -- Wrong prediction
"Deep learning models often prefer one dominant state. AF3 may over‑stabilize a closed form even when an open form exists"
6th-paragraph says -- Illusory protein structures
"AF3 reduces but does not eliminate the hallucination of structure in intrinsically disordered regions. Users should distrust low-confidence predictions."
middle-Training data bias says -- Trained on experimental data
"AF3 learned from the Protein Data Bank, which is skewed toward stable, soluble proteins. Membrane proteins, large complexes and disordered proteins are under‑represented, potentially limiting accuracy in these areas"
Even the latest Alphafold-3 (trained on the experimental static protein PDB data, this p.8 ) also cannot predict molecular motion nor protein conformational change ( this-2nd-last-paragraph ).
This-middle-Alphafold 3 model limitations says -- Useless static proteins
"AlphaFold 3 predicts only static structures, while dynamical behaviors of biomolecular systems are Not addressed"
This or this-lower-Limitations of Alphafold 3 -- Wrong Alphafold-3
"Like its predecessors, AlphaFold 3 produces static structures and does Not account for the dynamic behavior of molecules in solution, which remains a key limitation in protein structure prediction"
" AlphaFold 3 may produce incorrect or incomplete conformations for specific proteins"
So today's AI, Alphafold unable to clarify dynamical biological, enzymatic reactions are useless for discovering effective drugs or curing diseases.
Today's AI, Alphafold based only on experimental protein structures do Not understand the physical mechanisms of protein folding due to useless quantum mechanics.
This 2nd-paragraph says -- Mechanism unknown
"they (= Alphafold-3 ) cannot say why they are folded that way... How the AlphaFolds will catalyse drug discovery is also unclear."
This-p.8-right-first says -- Wrong Alphafold-3
"Although AF3 (= alphafold-3 ) has shown its potential ability to
detect mutation-induced structural changes, its accuracy remains inconsistent"
This-Alphafold3 overview-remaining challenges say
"Inaccurate structures in disordered regions, orphan proteins, highly dynamic proteins,"
"Produce overlapping atoms ( this-p.1-abstract )"
Furthermore, Alphafold-3 based on a generative model often gives illusory wrong protein structures called "hallucination ( this-Limitations, concern, this-1., this-the promise and challenge )".
This-7th-paragraph says Alphafold-3 is unreal
"it (= Aphafold3 ) does not support arbitrary chemical modifications, ligands, or ions, and it may generate non-existent secondary structures when uncertain, introducing a risk of hallucination (alpha helices instead of unpredicted regions)..
While it predicts only static structures and lacks modeling for conformational changes upon binding,"
This-3~5th paragraphs say -- Alphafold-3 wrong
"In some cases, the modelled conformational state may Not be correct"
"There are still some targets that AlphaFold 3 struggles to model. One challenging class is orphan proteins,"
"The updated model architecture means that AlphaFold 3 sometimes produces categories of error.. spurious structural order ( hallucinations, this-2nd-last-paragraph )"
Alphafold3 (= AF3 ) often failed to predict proteins different from its training PDB data, as this p.4-4th-pararaph says
"AF3 failed to produce the correct pose even in the unmutated case ( this-p.2-1st-paragraph )"
This-p.1-abstract-lower says -- Alphafold-3 failed
"However, AF3's (= Alphafold-3 ) 60%
failure rate for antibody and nanobody docking (when sampling a single seed = freely-adjustable input parameters )
leaves room for improvement to equip antibody design endeavors."
This-p.6-left-2nd-paragraph says -- Wrong Alphafold-3
"it is
concluded that AF3 complex prediction is Not reliable for
intrinsically flexible regions or intrinsically flexible domains of
PPI complexes"
Contrary to the media-hype, Alphafold-3 cannot predict structures of RNA or DNA ( this-p.6-challenges and limitations ).
This-1st~2nd-paragraph says -- Alphafold-3 wrong
"One of the biggest disappointments when AlphaFold 3 came out was that it wasn't very good at predicting the structures of nucleic acids."
This-p.12-left-4th-paragraph says -- Alphafold-3 fails
"AlphaFold3 does Not
reproduce all of the non-Watson–Crick interactions, which is
essential for the stability of 3D RNA structures. Furthermore,
AlphaFold3 fails to predict structures from orphan families"
This-p.12-right-2nd-paragraph says -- No drug discovery
"relying solely on AI prediction methods like AF3 (= Alphafold-3 ) is far from ideal or absolutely accurate.
".. AF3 still cannot meet the requirements in the field of drug design, and as its prediction ability means that it cannot replace experimental results"
This-lower-Conclusion (2025) says
"The tool (= Alphafold-3 ) isn't perfect. RNA remains challenging, dynamics are missing, and membrane proteins need work"
↑ Because today's experiments (= X-ray- crystallography, cryo-electron microscopes ) on which Alphafold are trained cannot get exact membrane protein structures (= modified by various other molecules ) at atomic level.
So AI, Alphafold cannot cure diseases.
(Fig.3) Alphafold-3 unable to predict protein motion has to rely on very old impractical molecular dynamics (= MD ) that is too time-consuming to simulate biological reactions. → drug discovery is impossible.
Today's AI, Alphafold trained on experimentally-obtained static protein database cannot predict any protein conformational change nor biological reactions.
So today's physics has to rely on the very old, extremely-time-consuming method called molecular dynamics (= MD ) to simulate molecular or protein conformational change ( this-5th-paragraph ).
The problem is the unphysical quantum mechanical atomic model and its density functional theory (= DFT ) lacking real atomic shape are impractical, unable to predict anything, and their calculations are unrealistically time-consuming.
This current unreal quantum mechanical shapeless atomic model hampers developing useful multi-probe atomic force microscopes by restricting the molecular-motion simulating method only to the impractical time-consuming MD.
Today's only simulating method = MD unable to treat molecules as real objects with shapes has to gradually move each (shapeless) atom based on artificially-created pseudo-potential called force field, which is too time-consuming and useless ( this-p.2-1 introduction ).
↑ This extremely time-consuming molecular dynamics (= MD ) also has to rely on experimentally-obtained proteins' structures (= wrong, unreliable ) in its artificial force field potentials that often fail, becaue the current mainstream useless quantum mechanics cannot predict any molecular behavior.
This recent research had to rely on this impractically-time-consuming MD simulation (= just 500ns protein-motion-change, which is useless, this-p.6-MD simulation) after Alphafold2 was used for giving static wrong protein structures. ← MD relying on today's wrong Alphafold's initial protein structures also gives wrong simulation.
This recent biological research also used only the too time-consuming MD (= just 500ns simulation, which is too short to simulate much-longer protein folding ) without using (useless) Alphafold ( this-p.10-MD simulation ).
Even the latest approximate accelerating MD methods can simulate only 8-μs protein's change ( this-p.7-3rd-paragraph ), which cannot simulate much-longer protein or biological reactions with seconds ~ hours time scales.
There are a few AI methods that try to predict some protein conformational change trained on experimentally-obtained protein structures.
↑ But initial protein structures given by Alphafold and MD's force field potentials heavily rely on today's limited wrong experimentally-obtained data, so AI, Alphafold can never simulate proteins' motion correctly.
This-Current limitations of Aphafold3, DiffDock,NeuralPlexer and DynamicBind says
"Only static structures are predicted (= in Alphafold-3 ).
Conformational changes upon binding are not modelled.
Hallucinations (= wrong protein prediction by Alphafold-3 ) are introduced, which is especially relevant for disordered proteins."
"Reaching the slowest transitions with classical MD (= too time-consuming molecular dynamics ) simulations can take months or require specialised expensive hardware like the Anton supercomputer. Using enhanced sampling techniques (presented in a future article), those transitions can be significantly accelerated but are still computationally expensive." ← MD is too time-consuming, impractical.
↑ These current leading AI methods (= DiffDock, DynamicBind.. ) cannot predict any new proteins that are Not included in their training datasets (= experimentally-obtained protein-ligand structures called PDBbind database was used for training these AI methods, this p.11-left-Data construction ).
This p.7-Figure 2 shows the success rates of predicting protein docking by all these AI or machine-leaning methods such as DiffDock, DynamicBind, NeuralPlexer get worse (= success rates are very bad, only less then 20% ) when new protein structure datasets called PoseBusters ( this-p.5-2.6.2 ) were used for testing them.
An AI method trained on the extremely-time-consuming MD results is also useless, too time-consuming to simulate actual proteins.
Another generative AI model called MDGen has to rely on this extremely-time-consuming MD simulating result database called ATLAS ( this-11th-paragraph, this-p.1-last-paragraph, p.5-2nd-paragraph ), so this MDGen cannot simulate practically-long protein reactions, either.
(Fig.A) Direct experimental observation of proteins by multi-probe atomic force microscopes is best, which is hampered by unreal quantum mechanics and MD model.
This recent hyped news (7/10/2025) says
"understanding how proteins function by shifting between different shapes in response to other molecules remains a central challenge...
Current molecular dynamics (MD) simulation... are slow, costly,.."
"This (= BioEmu AI method ) is accomplished by combining training data from AlphaFold structural predictions, large-scale MD simulations, and extensive experimental measurements of protein stability" ← AI needs experimental data and the impractical time-consuming MD data after all.
"BioEmu has limitations. For example, it does Not natively model molecular dynamics or interactions with membranes, ligands, or varying environmental conditions"
↑ So this new BioEmu AI method needs the extremely time-consuming MD and experimental data, hence it can Not predict mempbrane proteins lacking experimental data.
↑ Experimental observation is best. = Only multi-probe atomic force microscopes can directly observe and clarify detailed atomic mechanisms of all proteins, which is hampered by the unphysical quantum mechanical shapeless atomic model for 40 years.
↑ This research paper ↓
p.1-last~p.2 says -- Alphafold wrong
"AlphaFold and similar models have built upon the decades of data accumulated in the Protein Data Bank (PDB)... For protein function, unfortunately, methods that are both highly accurate and high-throughput
are missing" ← Alphafold cannot clarify proteins' functions or motions.
p.2-2nd-last-paragraph says -- Experiment needed
"We
therefore train BioEmu by combining data ranging from a large set of static protein structures and vast amounts
of (impractical) MD simulation to experimental measurements of protein stabilities"
p.6-2nd-paragraph says -- AI bad prediction
"predicts the
cryptic pocket in 85% of cases, while it only succeeds in predicting 49% of the apo structures, indicating further
room for improvement.
" ← This new AI method called BioEmu is also useless with bad predicting rates like other AI.
p.6-3rd-paragraph says -- Time-consuming MD
"Due to the vast computational
costs, exhaustive MD sampling of biomolecules has only been achieved in few cases" ← Molecular dynamics (= MD ) is impractical, too time-consuming to simulate proteins.
p.10-3rd-paragarph says -- Bad experimental data
"membrane environments and small molecule ligands are neither represented in the model nor in the
training, and BioEmu can therefore Not be expected to make reliable predictions when membranes or ligands,
play a key role in the process"
↑ This new AI or BioEmu is also useless, unable to simulate behavior of proteins whose experimental data are Not included in its training datasets (= so experimental observation instead of hyped AI is the most important ). ← Only practical multi-probe atomic force microscopes can directly observe and simulate all proteins' behavior in experiments.
(Fig.3') Overhyped AI, Alphafold, quantum mechanics are useless, Not utilized in actual medical researches.
AI, Alphafold are useless, unable to predict any protein conformational change, biological, enzymatic reactions.
Quantum mechanics, DFT and MD are also useless, too time-consuming to explain actual protein or biological reactions ( this-p.8-right-2nd-paragraph ).
Due to this useless quantum mechanical atomic theory, today's medical and biological researches are deadend, unable to consider the detailed atomic interactions nor clarify true biological mechanisms. ← curing diseases is impossible.
Today's medical researches just using macroscopic biological tools obtained from organisms such as bacteria do Not use the useless quantum mechanics (= Schrodinger equation, DFT ) nor the extremely time-consuming MD nor AI-Alphafold ( this-p.12-14-methods, this-p.9-12 this-p.10-14 ).
All these (impractical) quantum mechanical methods such as Schrodinger equations and DFT just choosing (fake) wavefunctions and exchange energy pseudo-potentials are unable to predict anything ( this-p.4-left-last-paragraph ), just hampering nano-technology.
(Fig.4) Today's experimental methods (= X-ray crystallography ), AI, Alphafold cannot clarify real protein structures or biological reactions.
It is impossible to know true protein, enzymatic conformational change and reaction only from the static protein structures (= PDB ) obtained from X-ray crystallography (= using crystallized static proteins ), NMR (= measuring only small proteins ), cryo-EM (= electron microscopy insufficient for drug development ).
Actual dynamical protein structures inside human bodies are often different from static protein structures (= such as X-ray crystallography ) registered in PDB database on which today's AI and Alphafold rely.
This is why all the current AI and Alphafold are unable to predict actual dynamical protein or enzymatic structures, functions.
The only possible way to know precise molecular or protein conformational change at an atomic level is atomic force (= AFM ) or scanning tunnel microscopes (= STM ).
The problem is today's atomic force or scanning tunnel microscope has only one probe tip that cannot know the target atom or grab individual atoms freely.
There is already a technology of atomic force or scanning tunnel microscopes with multiple probes, but these are used only for measuring electric conductance between two probes, Not for grabbing or identifying target atoms with multiple probes.
It is surprising that today's atomic force (or scanning tunneling ) microscopes still have only one probe tip, though more than 40 years have passed since they first succeeded in manipulating a single atom.
↑ This is due to the unrealistic quantum mechanical shapeless atomic model prohibiting researchers from dealing with atoms as real objects by developing multi-probe atomic force microscopes for a long time.
We should develop atomic force or scanning tunneling microscopes with multiple probe tips manipulating individual atoms freely (= this technology already exists, though ) as soon as possible to clarify precise proteins' behavior at atomic level and develop effective drugs for the current intractable diseases.
By using multiple probe tips (= to which single atoms are attached ), we can design and construct artificial molecular or protein devices in the bottom-up approach (= microscopes can manipulate molecular bond formation ).
In the same way, we can disassemble and analyze isolated natural proteins little by little by using multiple probes of atomic force or scanning tunneling microscopes (= by causing artificial hydrolysis, this or this-p.17-p.24 ) to know the precise protein structures inside human bodies in the top-down approach.
↑ By combining these top-down and bottom-up approaches using multi-probe atomic force microscopes, we can know and confirm the actual protein interaction, behavior and conformational change (= induced by approaching molecules or ions carried by microscope tips ), which leads to curing today's intractable diseases.
(Fig.5) Alphafold, AI, today's medical research cannot clarify true molecular mechanism nor find effective treatments due to useless quantum mechanics.
Today's overhyped AI, Alphafold and medical researches are unable to consider actual atomic interactions due to useless quantum mechanics.
This is why finding effective drugs for cancers and Alzheimer is still impossible despite long time researches (of AI or medicine ).
For example, the 5th, 7th paragraphs of this hyped news (10/27/2024) say
"Google DeepMind's artificial intelligence tool AlphaFold... to help them identify a new protein (= false, already-known protein ) that allows the first molecular connection between sperm and egg."
"Scientists still don't know how the sperm actually gets inside the egg after it attaches and hope to delve into that next." ← AI-Alphafold and biological researches are still unable to explain true mechanism of fertilization after all.
↑ This research paper ↓
p.2-1st-paragraph says -- Mechanism unknown
"our insights into vertebrate sperm-egg
binding and fusion lack mechanistic understanding" ← (useless) Quantum mechanics failed to clarify fertilization mechanism even 100 years after its foundation.
p.3-1st-paragraph says -- AI prediction wrong
" we performed an AlphaFold-Multimer in silico
screen against ~1400 zebrafish testis-expressed secreted/transmembrane proteins (= these are already-known proteins, Not new )"
"Intriguingly, Izumo1-Spaca6 was one of the top-scoring predicted interacting pairs, which was unexpected given that recombinant human IZUMO1 and SPACA6 ectodomains do not appear to interact in vitro" ← Alphafold's prediction failed.
p.3-2nd-paragraph says "AlphaFoldMultimer predicted with high confidence the formation of a trimer between zebrafish Izumo1, Spaca6, and Tmem81 (= new finding )"
p.5-2nd-paragraph says "we generated knockout lines in zebrafish and mice using CRISPR/Cas9.. Tmem81-/- male fish (= Tmem81 gene deleted ) and mice were sterile," ← Tmem81 protein seemed to be necessary for fertilization (= which was confirmed by biological experiments. Not by Alphafold ).
p.14-last-paragraph says -- Not AI prediction
"Tmem81 was independently discovered in a search for remote homologs
of Spaca6 proteins (= so this Tmem81 protein was already known to have homology with fertilization-related proteins before Alphafold )"
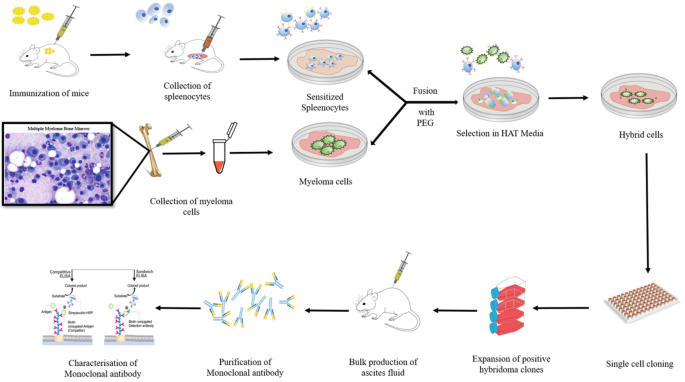
p.15~p.20 used conventional macroscopic biological experiments such as zebrafish/mouse fertility tests (using bacterial-CRISPR gene knock out ), immunoprecipitation based on antibodies (= obtained from immunized animals ), GFP fluorescent protein from jellyfish (= No quantum mechanics nor Schrodinger equation was used in this biological research ), without looking into detailed atomic interaction
This or this-p.10-Limitation of the study (= Discussion-lower ) admits
"we lack details
on stoichiometry and structure... Furthermore, our study does not provide in vivo evidence for complex formation... it is unclear whether
Tmem81's function extends beyond"
↑ Today's medical researches and AI-Alphafold had No ability to clarify atomic mechanism ( this 11th-paragraph ), so finding effective treatments for diseases is impossible, contrary to hypes.

Feel free to link to this site.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}