
トップページ (2電子原子も含む正確な新ボーア模型)
グルカゴンタンパク質構造のページに戻る。
ここでは タンパク質構造を調べるため 次のサンプル JAVA プログラムを使用する。
サンプル JAVA プログラム ( PDB タンパク質 )。
このプログラムは "ATOM" の部分だけ扱える ( "HETATM" や "CONNECT" の部分に関しては まだ未完成です。 )
グルカゴン以外のタンパクデータでも ある連続したアミノ酸 (= 連続した残基番号 ) のものを グルカゴンのデータ部分と置きかえれば このプログラムで扱える。
( ただし "N" (= 窒素 ) をスタート原子、として すべてのアミノ酸が完全にその原子を含んでいる必要がある。"OXT" は特に必要はない。また 自動的に水素付加するため 結晶解析データのみ扱える。 )
グルカゴンの PDB データ ( 1GCN ) は
このテキストは "C:\sdff" という名のフォルダーに "glucagon6.txt" というファイル名で保存する。
その後、上記の JAVA プログラムを "protein.java" としてセーブして コンパイルしてほしい。
”詳細は、-Xlint;uncheckedオプションを指定して再コンパイルしてください。”の 注意は無視して実行できる。
(Fig.1) プログラム内のフォルダー名の変更。
もし このテキストを別のフォルダーにセーブした場合は 上記のプログラム内の青線の部分を このセーブしたフォルダなどに変更するように。
例えば、"ATOM" (= アミノ酸 ) 部分のみ取り出した カルモジュリン ( PDB 1CLL ) テキスト もプログラムで実行できる。
1CLL は 140 アミノ酸以上から成るため スタートアップや アミノ酸鎖回転などに グルカゴン以上に時間がかかる。
(Fig.2) グルカゴン 3D 構造。

各原子間をつなぐ線は 灰色で 先端方向が 画面奥に向かっている (= x-y 平面では -z 方向 )。
(Fig.3) テキスト ( 左部分 ).

"*" のマークがついた原子は 現在選択している (= select ) 原子の意味である。
"-N-"、 "-CA-"、 "O"、 "C" は 主鎖にある 窒素、 炭素 A、炭素、酸素を表している。
"CB"、 "H0" は 炭素 (= 側鎖の最初 )、水素 ( 0 = N と結合、 1 = CA と結合、 4 = 側鎖の H ) である。
(Fig.4) 主鎖と側鎖。

基本的に タンパク質は 20種類のアミノ酸が結合して 主鎖と 側鎖に分けることができる。
主鎖は N-CA-C=O ( 窒素 - 炭素 A - 炭素 = 酸素 ) から成る。
そして 側鎖の最初の炭素が CB で 側鎖の他の原子は C', O', N', S' と ダッシュマークがついている。
X 線結晶解析の PDB データは 水素原子の情報が含まれていない。
そのため このプログラムは 各アミノ酸に自動的に水素を付加している。
原子番号は Fig.4 に示す順番についている ( 主鎖 → 側鎖 → 水素 → 主鎖 (次の) → 側鎖 .. )。
Fig,3 の2列目は "絶対的な" 原子番号で 実行中は 固定されており変わることはない。
(Fig.5) PDB データ形式。

"p1"、 "p2"、 "p3" は PDB フォーマット の原子番号 (= "3" ) である。
この PDB の原子番号は 水素原子を含んでいないため 上記の 絶対的な原子番号は 付加された水素分だけ大きくなる。
"PDB" ボタンをクリックすると、 2列目に "b1", "b2" ..., の番号が表示される。これは PDB の 残基番号である。
( この場合は ヒスチジンが "b1"、 セリンが "b2" である。 )
x, y, z は 絶対座標 ( Å ) である。
" dis (A) " は 各原子と 選択している原子 (= "select" ) 間の距離 ( Å ) である。
(Fig.6) 表示されている原子の変更.

"Atom" ボタンをクリックすると、 次の原子の列 ( 5 - 9 ) が表示される。
"+Z back-1" ボタンをクリックすると、1ページ前の列に戻る。
"+Y back" ボタンをクリックすると、 最初の原子の列 (= 0 - 4 ) に戻る。
"+X (A)" ボタンをクリックすると、選択した原子に対する相対座標が表示される。
"dis (A)" は 選択された原子と 各原子間の 距離である。
( このケースでは 0-N と 1-CA の距離は 1.473 Å である。 )
(Fig.7) 回転、半径、移動。

ある値 (= ° ) を "angle =" の隣のテキストに入れて、 "rotation" ボタンをクリックすると、 タンパク質全体が指定された平面上で回転する。
( x-y ang、 x-z ang、 y-z ang を選択する。 )
" distance = " の横のテキストに ある値 (= Å ) を入れて"center" ボタンをクリックすると、 "nucleus" 原子の周囲の 指定された距離範囲内の原子だけが表示される ( 最初の値は "80.0" Å である )。
( 例えば、 "4.0" を入力して "center" をクリックすると、 原子 "0" から 4.0 Å 半径以内の原子だけが表示される。 )
注意: この "nucleus" 原子と "select" 原子は別のものである。
"move" ボタンをクリックすると、 指定された距離 (= "distance" の隣 ) 原子全体が 移動する。
"x move"、"y move"、"z-move" などを予め選択しておく。
(Fig.8) 端のマークをクリックして 移動または回転させる。

三角マークをクリックすると、タンパク質全体が 指定された方向に移動する。
( x-y 平面の 上部の三角形をクリックすると、 タンパク質は y 方向へ移動する。)
移動距離は "distance =" 横の ( 正の ) 値 ( Å) である。.
( ここでは 単に距離の値をテキストボックスに入力して 何も他はクリックせず、三角ボタンだけクリックする。)
この値が 10 よりも大きいとき、移動距離は 2.0 になる。
端の 円 をクリックすると、指定された方向に タンパク質全体が回転する。
回転角は "angle =" の横のテキストボックス内の値を基にしている。
( テキストボックスにある角度の値を入力して ある円をクリックするだけでいい。)
最後の行の "angle" は 原子1− 原子2 (選択した) − 原子3 の角度である。
この原子1、原子3は 原子2と接したものである。
注意:この原子番号は Fig.3 の "2" の絶対的な原子番号であり、表示番号 (= 最初の列) や PDB の原子番号 (= p3 など ) とは異なるものである。
(Fig.9) 選択原子 (= "select" ) の変更。

"select" ボタンの横のテキストボックス内に原子番号を入力して select ボタンをクリックすると、選択原子の変更ができる。
この原子番号は "display" 隣の スクロールバー内の選択条件により 意味が異なる。
( スクロールバー内から条件を選択するだけで "display" ボタンはクリックする必要はない。)
"all" (= 初期状態 ) もしくは "number" が スクロールバー内で選択された状態で 入力された値は Fig.3 の "1" の"表示された" 原子番号である。
"atom" が スクロールバー内から選択された状態で入力した値は Fig.3 の "2" の 絶対的な原子番号である。
"PDBat" が選択された状態で入力した値は PDB の原子番号である。
( つまり このケースでは 水素は選択できない。)
(Fig.10) "nucleus" と "select" 原子。
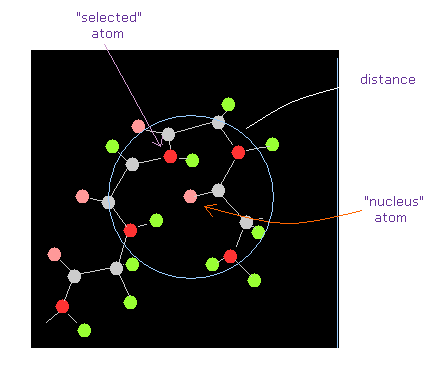
基本的に 計算値は "select" 原子との比較がなされる。
"nucleus" 原子は "distance" ( "dis (A)" でなく ) のみと関係している。
スクリーンに表示されている原子は この "nucleus" 原子から 指定の距離 (= Å ) 内のものである。
( 基本的にこのエリア内から ある原子を select する。)
この "nucleus" 原子を変更すると、新しい nucleus 原子が number 0 の表示番号となる。
つまり 上で触れたとおり、Fig.3 の "1" の原子番号は これらの操作で変わる。
( 一方、絶対番号と PDB 原子番号は 変化しない。)
もし 初期状態の表示番号に戻りたいときは、スクロールバーから "atom" を選択して "0" を "nucleus" 横のテキストボックスに入力して "nucleus" ボタンをクリックするといい。
すると 最初の窒素原子の表示番号が "0" に戻る。
(Fig.11) 画面上の原子を直接クリックして "select" 原子を変更。

画面上の ある原子を直接クリックして 選択原子を変更できる。
例えば、"46" の赤の円 (= 窒素) をクリックする。
この時点で、"to=" 横のテキストボックス内に "0" が入力されている状態だと 選択原子が "46" に変更される。
( "to=" ボタンはクリックする必要はない。ただ その横に "0" を入力するだけでいい。 )
もし "to=" 横に "0" 以外の数値を入力した状態で "46" 原子の丸をクリックすると、選択原子は変更されないまま、"46" 原子のテキスト画面にジャンプする。
( つまりこのケースでは "*" のマークが "46" 原子につかない。)
"select" 原子を変更せずに ある原子のテキスト画面に ただジャンプしたいときは、" to= " ボタンも使える。
"to=" ボタンの横に ある原子番号を入力して それをクリックすると その原子のテキスト画面にジャンプできる。
(Fig.12) 水素原子を含むすべてのアミノ酸を表示する。
プログラム実行後、主鎖、側鎖、水素原子すべてを含む原子全部が画面に表示される。
そのため 基本的なタンパク質構造を知りたいときは 例えば
" chain= " ボタンをクリックすると、 主鎖のみが 画面に表示される。
(Fig.13) 主鎖のみが表示される。

Fig.13 では、 主鎖のみが 画面に表示されている。
( このケースにおいても 表示番号の対応が変わるが、絶対番号などは変化しない。)
次に "hydrogen" ボタンをクリックする。
(Fig.14) 水素原子なしの主鎖のみが表示される。

"hydrogen off" の状態では 水素原子がタンパク質から除去される。
もちろん、この状態は 単に表示のためだけのものである。
ポテンシャルエネルギーなどの相互作用計算は この表示状態でも 全部の原子の効果について計算される。
" chain= " ボタンをもう一回クリックすると、側鎖のみが表示される。
そして " chain= " ボタンを もう一度クリックすると、 "all" (= すべて ) の原子表示に戻れる。
(Fig.15) 表示される原子 ( アミノ酸 ) の選択。

スクロールバー内から 原子番号条件 ( 例えば "number" ) を選択して、" from(dire)= " と " to= " の横のテキストボックス内に 原子番号を入力して
"display" ボタンをクリックする。
Fig15 の左では、 19 から 48 の原子のみが表示される。
( 例えば、 "N" を " from " 原子として選択すると、スタート地点のアミノ酸のすべての原子が含まれる。 )
スクロールバー内から "residue" を選んで PDB の残基番号を入力して "display" ボタンをクリックする。
Fig.15 右では、 "2" と "3" の残基のみが画面上に表示される。
もし 1つのアミノ酸のみを表示したいときは 例えば "from" と "to" の テキストボックスに "2" と "2" などと入力すればいい。
(Fig.16) タンパク質全体を ある方向へ向ける。

スクロールバー内から "atom"、 "number"、 "PDBat" の条件を選択して、 ある 原子番号を " from (dire)= " の横のテキストに入力し、下の スクロールバー内から 方向を選択 ( x, y, z, -x, -y, -z ) して"turn atom" ボタンをクリックする。
Fig.16 のケースでは、 タンパク質全体が 5 → 6 原子 (= "表示" 原子番号 ) のベクトルを -z 方向に向けるように回転する。
この操作は 二面角の確かめなどに使うことができる。
(Fig.17) 二面 ( ねじれ ) 角。

二面角は この情報が分かれば タンパク質の 3次元構造を組み立てることができるため 非常に重要な概念である。
二面角は Fig.17 で 平面 A (= 29-30-31 ) と 平面 B ( 30-31-46 ) のなす角である。
"from (dire)=" ボタンの隣に "31" を入力して ( Fig.17 では 絶対原子番号条件を選択している ) 、 "dihedral" ボタンをクリックすると、この二面角を得ることができる。
二面角では 水素原子や酸素原子などは 関係していないことに注意する必要がある。
( N, CA, C もしくは CB ) を選択した後 二面角を得ることができる。
( 基本的に ペプチド結合は 2重結合性のため "N" を選んだ場合 この角度は 約 -180°になる。 )
(Fig.18) 2面角と 原子の選択。

例えば、平面 0-1-2 と 平面 1-2-18 間の角度を知りたいときは、 原子番号 "2" を テキストボックス内に入力して、 "dihedral" ボタンをクリックする。
基本的に CA や C の炭素原子直前の角度は ラマチャンドラン・プロット として知られており、αへリックスや βシートの構造などで 非常に重要である。
もし N ( 例えば "18" ) を テキストボックス内に入力すると、 角度 CA-C-N-CA は 約 -180°であることがわかる。なぜなら ペプチド結合に含まれる原子は その2重結合性のために ほぼ 同一平面内にあるからである。
"4" (=CB ) を入力すると、 平面 0-1-4 と 平面 1-4-5 の間の角度を知ることができる。
(Fig.19) 二面角と原子位置。

Fig.19 に示したように、 29 と 46 が "cis" の位置に配置されているとき、 二面角は "0"°になる。
一方 それらが トランスの位置関係にあるとき、二面角は -180° ( もしくは 180° ) になる。
Fig.19 (= arrow 30 → 31 ) の視点からみて、 ベクトル 31-46 が 反時計まわりに ある角度 ( 例えば、30° ) 回転すると、二面角は マイナス ( -30° ) になる。
一方、時計周りに回ると、その角度は プラスになる。
( Fig.17 のケースでは、 この角度は -12°になる。 )
(Fig.20) "C" の炭素部分での回転。

次に アミノ酸鎖の一部を回転して ポテンシャルエネルギーや力などが どのように変化するか調べることにする。
基本的に 回転のスタート地点のアミノ酸は "CA"、 "C" もしくは "CB" になる必要がある。
そのため Fig.20 では、 "2" (= C ), "4" (= CB ), 19 (= "CA" ), 22 (="CB" ), "20" (= C ) を (絶対)原子番号として選べる。
( 原子番号 "1" も CA だが、 端の位置のため この原子選べない。 )
Fig.20 では、 ( 絶対 ) 原子番号 "2" と "20" を "from (dire)=" と " to= " テキストボックス内に入れる。 ( 入力するだけで "display" ボタンはクリックする必要はない。 )
それから "30" (°) を "angle=" の横に入力して、真ん中あたりの"chain rot" ボタンをクリックする。
( 左側の "rotation" ボタンはクリックする必要はない。 )
"C" を スタート地点の原子として選ぶと、 最初のアミノ酸 (= HIS ) 内では "C" (= 2 ) と "O" (= 3 ) のみが回転する。
"to=" 原子を含む最後のアミノ酸 (= SER ) は 全体が回転する。
つまり、多角形内に含まれる原子全体が 1-2 (= CA-C ) を軸として 30°回転する。
結果的に "2" の二面角は
-159.3° から -129.3°に変化する。確かめてみてほしい。
SER 内で 他の原子 ( 例えば "19" や "22" ) などを "to=" 原子として選んでも 回転する範囲は 同じである。
(Fig.21) "CA" 炭素における アミノ酸鎖回転。

"CA" の炭素を 回転スタート原子として選ぶと、 "N" と "H0" を 除いた 最初のアミノ酸 (= SER ) 内の すべての原子が 回転する。
グルカゴンは 470 の原子を含む。
そのため 例えば "500" を "to=" のテキストボックス内にいれると、 CA (= 19 ) の後すべての原子を 回転させることができる。 ( Fig.21 では、 -40° ).
(Fig.22) "CB" (= 側鎖 ) における回転。

側鎖の炭素 "CB" の原子番号を 入力したときは "to=" の原子番号は 任意のものが得られる。
例えば、 "5" もしくは "27" を "to=" のテキストボックス内に入れても、ヒスチジンの側鎖全体が回転する結果は変わらない。
"CB" (= ""4 ) の二面角は 平面 A (= 0-1-4 ) と 平面 B (= 1-4-5 ) の間の角度である。
" =back " ボタンを押すと、元のの無回転状態に戻れる。
(Fig.23) 窒素における固定された相互作用部分。

ここでは アミノ酸鎖回転後の エネルギーや力の 変化 が知りたい。
Fig.23 の右図では、原子が N-CA 軸周囲を回転したとき、 N-CA、 N-CB、 N-H 間の距離は 変化しない。
さらに C-N のペプチド結合は 安定な平面を形成する。
これらの事実を考慮して、 N 原子と 線で囲まれた部分の原子間の相互作用は考慮しない。
なぜなら 共有結合は 基本的には 他の力よりも はるかに強く安定であるからである。
(Fig.24) CA 原子における 固定された相互作用部分。

同様に、Fig.23 の固定されたペプチド結合を考慮すると、CA と Fig.24 の線内の相互作用を計算する必要がない。
また ここでは 5.0 Å 以上離れた原子間の相互作用は無視した。
分子動力学 ( MD ) 法では、すべての粒子間のクーロン相互作用計算に多大な時間がかかる。
量子化学が 大きな分子に適用できないため、MD 法では 具体的な電子分布という概念が存在しない。
つまり MD における クーロン相互作用というのは 正の原子核間やイオン間のクーロン力を意味している。
(Fig.25) タンパク質結晶は 濃い溶液や 他のタンパク質に囲まれている。.

MD 法とは異なり、ここでは リアルな電子分布を使用している。
そのため 負の電子が 正の原子核をキャンセルできるため 遠くの原子の影響力は無視できる。
タンパク質結晶を作るには たくさんのタンパク質を凝集させる非常に特殊な溶液の条件を選ぶ必要がある。
これはつまり これらの溶液は アミノ酸自身よりも強い力を発揮する 様々な塩やバッファーを含んでいることになる。
遠くの原子は 小さな回転でも 移動距離が大きくなる。そのため これら 塩や周囲のタンパク質が この空間に入り込んできて その変化を打ち消すことになる。
(Fig.26) 各アミノ酸構造は ドブロイ波長によって決まる。

ここでは このページや このページに示した電子分布状態を使う。
炭素原子周囲の広い空空間やその電子の周期的運動を考慮して 平均の電子分布状態を使う。
( C では -e × 4 の電子の代わりに -0.5e × 8 の電子を用いる。)
これらの原子半径は クーロン力やドブロイ波長を用いて得ることが可能である。
重要な点は 現在の量子化学や分子動力学法は これらの現実的かつ非常にシンプルな電子分布を使用できないことである。
(Fig.27) 量子力学の広がった電子雲 → 全電荷 = ゼロ !?

実は Fgi.26 の 現実的に分離した電子分布というのが 古典軌道による分子結合に関して 重要な鍵である。
量子力学や化学は この現実的で"分離した"電子の概念を使用できない。なぜなら ボーア模型に置き換わった量子力学の波動関数は滑らかに分布しているからである。
しかしもし 電子が量子力学のように あらゆる場所に細かく分離して広がっていると 例えば 電子軌道半径の違いなどがなくなってしまう。
なぜなら ご存じのとおり、無限小の電荷 (電子) が ある球面上に広がって分布しているとき、そのポテンシャルエネルギー V は この球がいかなる半径でも同じになってしまう。
( この ポテンシャル V は 球の中心からの距離のみの関数である。)
そのため 例えば、フッ素と塩素の違いが
なくなってしまい、同じ量のプラスとマイナスの電荷がちょうど打ち消しあって 常に 全電荷がゼロになってしまう。これは非常に奇妙である。
(Fig.28) 平均の水素の電子分布。

水素原子は たった1つの電子しかないため その平均の電子分布を考えることは 非常に重要である。
ここでは 水素原子に関しては 結合軸に垂直な平面上に -0.25e の4つの電子が分布している状態を考える。
そして その電子軌道が少し収縮していることを考慮して その半径を 0.4500 Å とする。
(Fig.29) 力 = 1000.
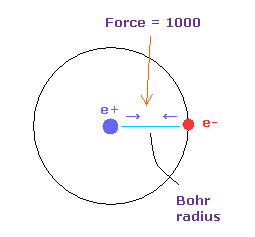
力の単位は Fig.29 に示したものを使う。
水素原子の基底状態で 電子 と +e の原子核間の距離が ボーア半径のとき、その力を 1000 とする。
例えば 1468 の力は H 原子の基底状態のときの力の 1.468 倍ということになる。
(Fig.30) 各原子に作用する力。

FX、 FY、 FZ は 他の原子核や電子から 各原子核に作用する力である。
( ここでは FX = 29.9, FY = -164.2, FZ = 72.3 になっている。 )
"all" のマークは グルカゴンタンパク質すべてにわたって計算したという意味で これがノーマルの状態である。.
"alv" は この原子が 5.0 Å 以内の範囲の電子や原子核全体と相互作用した際のポテンシャルエネルギーである。
( これらの力や V は Fig.23 や Fig.24 に示したような 各原子に近い 固定範囲は除外している。)
"FZ totF" ボタンをクリックすると、 (FX, FY, FZ) のトータルな力が "FX totF" のテキストボックスのところに表示される。
もう一度 それをクリックすると 元の状態に戻れる。
(Fig.31) 選択した原子に作用する力。

"FX rela" ボタンをクリックすると、選択原子と 他の様々な原子間の相互作用を知ることができる。
Fig.31 では 原子番号 "0" が選ばれているため、例えば 3-O-HIS の行の "rel 45.3" は 3-O-HIS (= 酸素 ) が 0-N-HIS 原子 (= 窒素 ) に作用する力を意味している。
また 0-N と 3-O 間のポテンシャルエネルギーは 0.3056 eV であることを示している。
この状態では すべての情報は 選択原子に対する 相対的な値となる。
そのため "2.99" は 3-O-HIS と 0-N-HIS (= 選択原子 ) 間の距離が 2.99 Å であることを意味している。
1-CA-HIS、 2-C-HIS、 4-CB-HIS 原子は Fig.23 に示したように 0-N-HIS 近くの固定エリアに含まれている。
そのため ここでは それらとの相互作用は計算していない。
(Fig.32) 選択原子に作用する力 - ページ2

左上の "Atom" ボタンをクリックすると、次のページに進める。
例えば 5-C'-HIS (= 側鎖の炭素の1つ ) の 0-N-HIS (= 選択原子 ) に作用する力は 33.7、 それら2つの原子間の V は 0.2727 eV である。
"FX rela" ボタンをもう一度クリックすると、元のオリジナルのページ (= 選択原子と比較しない ) に戻れて、5-C'-HIS そのものの 全ポテンシャルエネルギーは 0.8027 eV となる。
Fig.23 や Fig.24 に示した 無視された固定エリアは 同じ側鎖においても 原子位置に応じて異なるため トータル V は それらで少し異なる。
(Fig.33) 選択原子に作用する力 - ページ3.

もう一度 "Atom" ボタンをクリックして さらに次のページに進む。
一目見て、水素の (反発)ポテンシャルエネルギーが 非常に小さいことが分かる。
例えば、10-H0-HIS (= ヒスチジンの N に結合 ) のポテンシャルエネルギー V は マイナス (= -0.0158 eV ) である。
他の水素原子においても 反発相互作用は Fig.32 ( alV ) の他の原子と比較して非常に小さい。
水素原子の 選択原子に作用する反発力も Fig.32 の他原子に比べて非常に小さい。
14-H4-HIS (= 側鎖の水素の1つ ) の場合、相対的な力が マイナスになっており、これは この水素が N 原子を 引きつけていることを示している。
お気づきのとおり、水素原子の これら非常に小さな反発力は 水素結合などの 本当のメカニズムを表している。
( 10-12 の水素原子は N 近くの固定範囲に含まれているため これらの相互作用は計算されていない。)
(Fig.34) C (= "2" ) における アミノ酸鎖回転後の 力の変化。

ここで 2-C-HIS において 10°アミノ酸鎖を回転させる。
2-C 炭素の後のすべてのアミノ酸を回転させたいため "500" (= マックスの原子の数 ) を "to=" のテキストボックス、
"2" を "from (dire)=" のテキストボックスに入力して "chain rot" ボタンをクリックする。
アミノ酸鎖回転後は すべての相互作用を計算するのに少し時間がかかる。
この回転後、二面角は -159.3° から -149.3°に変化する。 ( 確かめるといい。 )
" =back " ボタンをクリックすると、回転前の元のタンパク質構造に戻る。
(Fig.35) N と O 間の距離は C (= "2" ) での回転後 長くなる。

最初の 二面角が -159.3° ( N-CA-C と CA-C-N の二面の間の角 ) のとき、 0-窒素 と 3-酸素 は ほぼ "cis" の位置にある。
"2" の位置で 10°回転後、 この酸素は 窒素から少し離れる。
この距離の変化は " V change " ボタンを押せば確認できる。
"cha" は 各原子と選択原子間の距離 (= O-N ) の変化 の意味である。
O-N の距離は 2.99 Å から 3.065 Å に少し長くなるため、 この変化量は 0.0755 Å (= cha ) ということになる。
また " c,aV " は この酸素のポテンシャルエネルギーの変化 (eV) を意味しており、 O-N 長が長くなるため、エネルギーは低くなる。
他の "c,al" などのパラメーターは 各原子に作用する力の変化である。
(Fig.36) O による反発力は C (= "2" ) における アミノ酸回転で弱くなる。

"FX rela" ボタンをクリックすると、各原子と選択原子間の 力やポテンシャルエネルギーが どのように変化したかが分かる。
"c,rl" = -2.0 は、 3-O-HIS の 0-N-HIS に対する反発力が 2.0 減った ( ここでは 45.2 → 43.2 ) という意味である。
これらの原子間のポテンシャルエネルギーは 0.0055 eV 減る ( ここでは 0.3056 eV → 0.3001 eV )。
なぜなら これらの間の距離が長くなったからである。
(Fig.37) コマンドプロンプト。

アミノ酸鎖回転後、結果の一部がコマンドプロンプト画面に表示される。
基本的に 非共有結合は 反発力が増強するため 許される最短の距離というものが存在すると考えられる。
Fig.27 の最初の行では、 HIS-CB-4 (= ヒスチヂン側鎖、 炭素 ) と SER-N-18 (= セリン主鎖、窒素 ) 間の距離が 短くなりすぎる ( 2.5407 Å → 2.4867 Å )。
この分子結合長は 立体障害を考慮すると 短すぎると考えられる。
"Energy change = 0.5470 (eV) " は このアミノ酸鎖回転により、全ポテンシャルエネルギーが 高くなることを表している。
つまり この回転は起こりにくく、元のタンパク質構造が安定であることを意味している。
( -10°回転においても、全ポテンシャルエネルギーが大きくなり、 これはつまり 元の位置が 安定であることを意味している。 )
また Plus のセクションの " 5= 0.0103 " は 原子 "5" (= 絶対原子番号 ) のポテンシャルエネルギーが 0.0103 eV 上昇することを意味している。
これらのセクションでは そのポテンシャルエネルギーの変化が特に大きくなった (= Plus ) もしくは 減った (= Minus ) 原子のみを表示している。
最後の行の "angle" では、例えば " 10-n-11 109 " は 原子"10"- n (= "選択" 原子 ) - 原子 "11" 間の角度が 109° ( ここでは "選択原子" は 0-N-HIS ) であることを意味している。
これらの原子番号は 2列目の 絶対原子番号である。
原子 10 と 11 は 窒素-0 に結合した水素原子 (= H0 ) である。
"zoom" ボタンの横に何か値を入力して ボタンをクリックすると タンパク質のズームが変化する。
"100" は 元の値で、もし "200" を入力して それをクリックすると、タンパク質の画面は 2倍に拡大する。
"distance=" の横に何か値 ( Å )を入力して "center=" ボタンをクリックすると、"nucleus" 原子周囲の 指定の半径内の原子のみ表示される。
( 注意:この 中心は "nucleus" 原子で "select" 原子ではない。)
このボタンをクリックすると、画面に表示されている平面が変化する。
"FY near button" をクリックすると、 このプログラムは 画面に表示されている原子間でのみ ポテンシャルや力を計算してだす。
( もとの状態では グルカゴン全体の原子に関して計算している。)
もう一度クリックすると、元の値に戻れる。
"inversion" ボタンをクリックすると、選択原子内で 電子の分布が逆になる。
( つまり もともと対称的な分布だと変化しない。)
"distance =" ボタン横に値を入力して "nuc" ボタンをクリックすると、選択原子内で 電子軌道半径が変化する。
"charge Z" ボタンをクリックすると、選択原子の中心電荷が変化する。
スクロールバー内から 何かの原子を選択して "element" ボタンをクリックすると、選択原子の種類が変化する。

2013/7/12 updated This site is link free.