
Home page
Quantum computing is useless
Quantum computer cannot simulate materials
(Fig.1) IBM error-prone useless quantum computer could only randomly sample fake trial wavefunctions chosen beforehand, which cannot predict anything nor discover drugs.
The recent overhyped media falsely insisted that IBM-Cleveland clinic-Riken quantum computer could simulate (= Not predict ) the largest 12635-atom protein, taking 100 hours (= impractical ), which may lead to useful drug discovery someday (= never happen ).
↑ The point is that they did Not say quantum mechanics or quantum computers could predict atomic energies, instead, they carefully chose the phrases such as "quantum computers could simulate molecules" and "model atomic behavior".
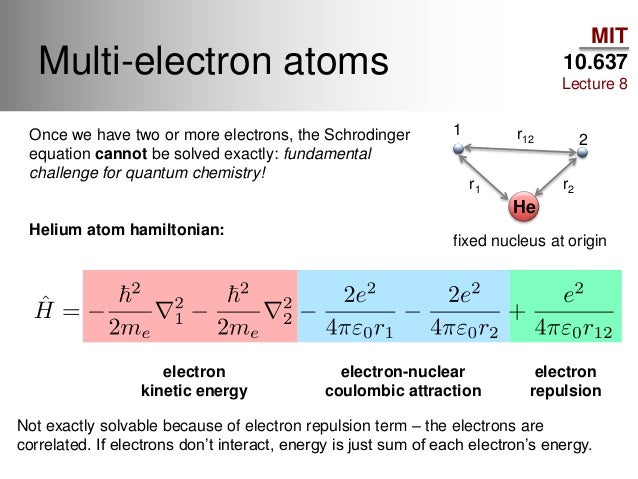
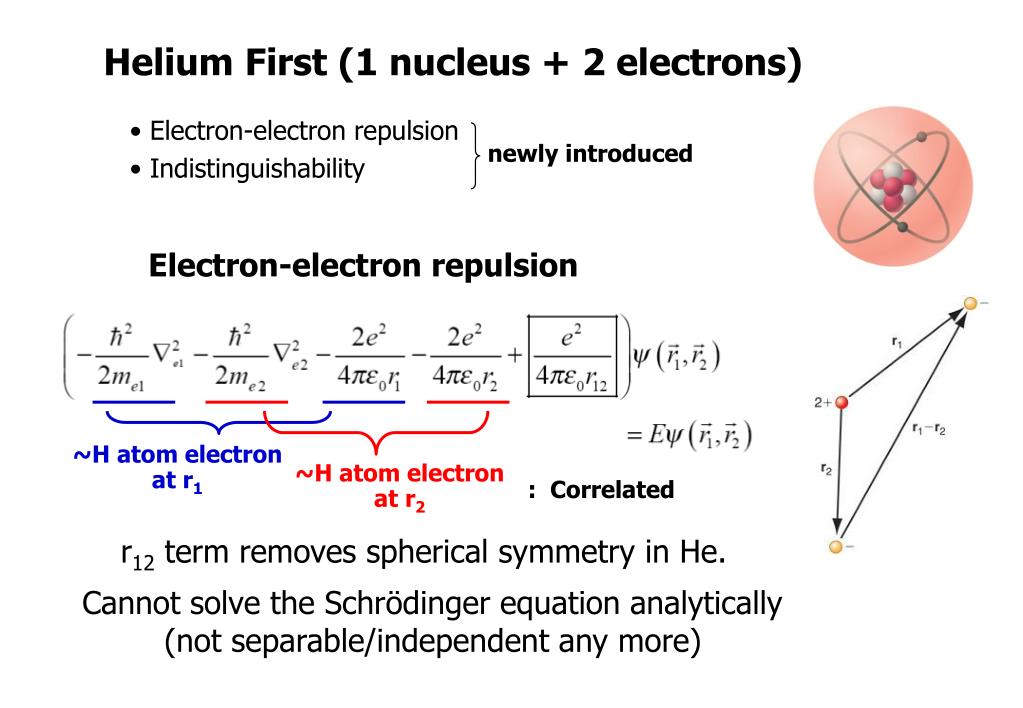
Because researchers know the old unrealistic quantum mechanics cannot solve its Schrödinger equation nor predict any atomic energies except for one-electron hydrogen atom, and today's error-prone parallel-world quantum computers cannot factor even the simple 21.
This or this-p.1-2nd-last-paragraph says -- Quantum mechanics impractical
"QM (= quantum mechanical ) methods apply the laws of QM to approximate the
wave function and to solve the Schrödinger equation. However, the Schrödinger equation cannot actually be solved for any but a
one-electron system (the hydrogen atom), and approximations need to be made ( This-p.4-left-last-paragraph )".
This or this-4th-last-paragraph says -- No quantum drug discovery
"For systems larger than a few particles, this becomes impossible really really quickly.... But even now, the strongest of computers, cannot analyze quantum systems larger than a few dozen atoms."
Quantum mechanics cannot solve its Schrödinger equations nor predict any multi-electron atomic energies ( this-or this-12th-paragraph ).
So physicists have to choose fake trial antisymmetric wavefunctions (= basis sets, ansatz ) or Slater determinants (= fake solutions ) out of infinite choices and infinite parameters ( this-4~5th-paragraphs ) to find ones giving the lowest energy in the impractical quantum approximate variational methods such as Hartree-Fock and configuration interaction (= CI or full-CI ), which are too time-consuming, can not predict anything ( this-p.6-2.2~p.7, this-p.6-lower, this-9.80 ).
This or this-p.4-1st-paragraph says -- No quantum mechanical prediction
"So we do not have any way of
knowing how good or how bad our trial wave function is.
The variational method also does not tell us what kind of a trial function we must choose"
This IBM research used deceptive hybrid quantum-classical methods called Sample-based quantum diagonalization (= SQD, this-12~15th-paragraphs, this-8th-paragraph ) which is equal to quantum selected configuration interaction (= QSCI, this-p.25-p.27 ) which quantum approximate variational methods cannot predict atomic energies due to the unsolvable quantum Schrodinger equations unable to predict anything ( this-figure-middle, this or this-p.4-1st-paragraph ).
↑ This IBM hybrid quantum SQD = QSCI method just artificially selects (fake) trial wavefunctions or unphysical Slater determinants ( step-4-(64), this-p.2-left ) for unsolvable Schrödinger equations, hence, its results depend on the artificial input values with No quantum mechanical prediction.
Today's error-prone useless quantum computers are used only to randomly sample or pick trial wavefunctions (= called configurations or Slater determinants, this or this-p.3-Fig.1 ) from trial wavefunctions (= called ansatz ) already chosen in advance without quantum computation (= and a classical computer has to calculate all values and energy ), so quantum computers are unnecessary.
This-p.16-left-4~5th-paragraphs say -- No quantum computer prediction
"We remark that the performance of QSCI (= SQD ) depends
highly on the quality of the input state."
"we proposed QSCI, a class of hybrid quantum-classical algorithms, to find low-lying eigenvalues and eigenstates of a many-electron Hamiltonian. Taking rough (= Not exact ) approximations of such eigenstates as input, QSCI selects (= Not predict ) important electron configurations to represent the eigenstates by sampling the input states on quantum computers, and then classically diagonalizes the Hamiltonian (= classical computer must calculate total energy in useless quantum mechanics that cannot predict anything, this or this-lower-equation 56 )"
In this IBM research, for unsolvable Schrodinger equations, first they chose fake trial antisymmetric wavefunctions (= IBM used simple inaccurate minimal basis sets, this-p.6-right-2nd-paragraph = minimal basis set, this-p.14-Some notions = minimal basis set is a poor choice ) whose (fake) atomic energies must be calculated by classical computers with No quantum mechanical prediction.
↑ Calculating all orbitals of 12635 atoms was too time-consuming and impossible, so IBM had to calculate only some chosen (= trimmed ) orbitals, determinants (= fragments, this-p.5-right~p.6-left ), which means this research gave up obtaining the exact energy of 12635 atoms from the beginning.
Their useless error-prone quantum computer was used only to randomly sample or pick orbitals for (unphysical) Slater determinants out of these artificially-chosen (fake) trial wavefunctions (= fragments, this-p.5-Fig.2 ) called ansatz (= this-p.3-(1) ) whose energy was estimated by a classical computer (= using impractical quantum approximations such as Hartree-Fock, CCSD, MP2.. ) in advance ( this-p.2-A, this-p.2-last, this-Fig.middle ).
This or this-last-6.Summary says -- Useless quantum computer
"In SQD, a quantum computer generate samples and a classical computer projects a Hamiltonian (= total energy ) onto a subspace spanned by the samples and diagonalizes it to compute eigenvalues and eigenvectors."
"The generated samples should be from the target (ground) state support." ← So quantum computers are meaningless, can only sample or pick trial wavefunctions from the already-prepared ground-state trial wavefunctions
This-p.3-2nd~3rd-paragraphs say -- Just choosing input wavefunctions
"The quality of the approximate energies and eigenstates obtained by QSCI (= SQD ) is determined by the choice of the input
state |Φ⟩ (= chosen trial wavefunction ). The input state must contain important bases to express the exact ground state with large weights so that
those bases appear frequently in the measurement results and are picked up in the selected CI calculation. One of the
ideal candidates for such an input state is the exact ground state" ← So just choosing the exact input trial wavefunction in advance with No quantum prediction.
↑ So quantum computer must (randomly) sample orbitals (= Slater determinants ) from trial wavefunctions artificially chosen to be close to ground-state in advance without quantum prediction.
Then, the classical computer must recover the originally-chosen trial wavefunctions from the noisy quantum computer-sampled orbitals (= configuration recovery ) and calculate total energy (= Hamiltonian diagonalization, this-p.5-p.6, this or this-Background, this or this-lower-equation-56 ).
↑ Diagnalization is calculating coefficient parameters giving the lowest energy in chosen trial wavefunctions ( this or this-7.10, this or this-p.5-2.4 ), which must be done by a classical computer (= today's error-prone quantum computers cannot calculate anything ).
This or this-4th-paragraph says -- Classical computer needed
"The technique underlying this result is Sample-based Quantum Diagonalization (SQD), the same method IBM has been developing as the flagship application for its QCSC architecture. In this workflow, a quantum processor (randomly) generates samples representing electronic configurations (= Slater determinants). Classical supercomputers then take those samples through configuration recovery, subsampling, and subspace diagonalization to estimate ground-state energies."
↑ So quantum computer's random sampling is unneeded. Just classical computers directly calculating energy from the artificially-chosen trial wavefunctions (= ansatz ) are OK ↓
↑ IBM quantum computer just randomly sampling was useless and meaningless. Just the classical computer was needed to calculate (= Not predict ) atomic energy from artificially-chosen fake trial wavefunctions in this research, contrary to hypes ↓.
This or this-review in the previous IBM SQD paper admits quantum computers are unneeded. ↓
p.2-last~p.3 says -- Quantum computer unneeded
"(i) a classical calculation ( of energy of chosen trial wavefunction by classical computers using quantum approximate CCSD or MP2 ) that (ii) informs
the choice of the parameters in the LUCJ wavefunction prepared on the quantum hardware. Then (iii) batches of bitstrings x (= electronic configurations) are measured (= sampled ) from such ( quantum ) hardware and (iv) such bitstrings are used to setup sub-spaces onto which (v) the Hamiltonian is projected and then classically
diagonalized (= a classical computer calculated atomic energy ).."
" The quantum part of the algorithm (iii) is therefore used to extract (= sample ) electronic configurations with a probability related to how much they were represented in the initial ( chosen ) wavefunction, that in turn was informed by the initial classical calculation. I think it should be made clear what is the practical advantage in extracting such configurations from the quantum hardware rather than directly from the initial classical wavefunction"
p.32-1st-paragraph says -- Quantum computer sampling meaningless
"In particular it is still Not clear if the SQD leads to a more
efficient sampling of the wavefunction w.r.t the classical approach"
↑ So directly using the chosen trial wavefunctions for atomic energy calculation by classical computers is OK by omitting the process of the quantum computer's randomly sampling numbers from the chosen trial wavefunctions.
This IBM research, which ignore some atomic orbitals and energies due to time constraints, could Not give exact atomic energies, hence they did Not compare their ( inaccurate ) calculated energies with experimental energies of 12635 atoms, hence, the phrase of "simulating atoms" is false. Drug discovery is definitely impossible.
Instead, this IBM research just compared their inaccurate hybrid quantum results with some classical computer results (= which were also wrong due to inaccurately chosen fake trial wavefunctions ), and showed No quantum computer advantage nor utility.
This-IBM-paper-p.10-right-2nd-paragraph says -- Wrong quantum results
"As a result, we cross the 12,000-atom barrier and perform
fragment calculations with accuracy higher than previously
conducted using HQC (= quantum-classical ) methods and matching leading classical
WF (= wavefunction ) method (= Not matching experimental results nor showing quantum advantage )" ← They just compared their inaccurate quantum results with some classical computer method, Not with experimental atomic energies.
This or this-8th-last~7th-last-paragraphs say -- No quantum computer advantage
"The work does Not yet outperform the best classical chemistry methods across all categories ( this or this-5th-last-paragraph )"
↑ This IBM research paper (= this-12th-paragraph or this-6th-paragraph-link ) ↓
p.1-right-last-paragraph says -- Quantum mechanical DFT fails
"The primary challenge towards this goal is to solve the
computationally costly quantum-mechanical many-electron
Schrodinger equation (SE) with accurate and scalable methods.
This task typically involves density functional theory (DFT)... DFT offers
more scalable solutions, but is based on semiempirical approximations that may Not produce accurate results
and lead to false positives and"
p.2-right-4th-paragraph says -- Quantum mechanics is impractical
"full configuration
interaction method (FCI = impractical quantum variational approximation just choosing fake wavefunctions or Slater determinants for unsolvable Schrodinger equation that cannot predict anything, this-p.3-3,p.7-5-Caveat ) are restricted to small and chemically
unrealistic contexts. Consequently, practical methods introduce
approximations ( this-p.2-introduction = giving up exact energy )"
p.3-left-1st-paragraph says -- Select wavefunction, No prediction
"The accuracy of SQD (= sampled based quantum diagonalization = quantum selected configuration interaction QSCI = deceptive hybrid quantum-classical method ) is
systematically improvable by increasing the number of selected configurations (= useless quantum computer can only randomly sample or choose fake trial wavefunctions or Slater determinants, which cannot predict exact atomic energy )"
p.3-right-last says -- Quantum SQD failed
"SQD
and ExtSQD may yield inaccurate results,"
p.4-left-1) says -- Classical Hartree-Fock calculation
"Given the geometry of a molecule we perform a Hartree Fock calculation (= inaccurate quantum approximation calculated by a classical computer ),.. to generate a set of molecular orbitals that are the input of quantum embedding"
p.5-Fig.2 shows --
Quantum computer unneeded
This useless IBM quantum computer was used only to sample trial wavefunctions (= unphysical Slater determinant parameters ). All atomic energy calculations such as configuration recovery (= generating usable trial wavefunctions from noisy quantum computer's sampling ) and diagonalization (= calculate
energy and coefficients, this-(61), this-p.5-(2.4) ) were conducted by a classical computer in this hybrid quantum method = SQD = QSCI ( this-p.4-1st-paragraph ).
p.6-right-2nd-paragraph says -- Choose inaccurate basis function
"For each complex, we obtain an initial geometry from
a PDB crystal structure (= using experimental data with No quantum prediction )..
We perform all quantum-mechanical calculations with the
STO-3G minimal basis set (= minimal basis set is the most inaccurate basis sets chosen for unsolvable Schrodinger equations that cannot predict anything, this-p.14-Some notions )"
p.8-left-A results say -- Inaccurate results
"EWF-TrimSQD (= this quantum hybrid method ) yield binding energies that
are comparable but too positive (= inaccurate )... the use of a minimal STO-3G basis set and the
values of η chosen to design fragments are likely causes for
this observation. Future work will (= still unrealized ) be directed at testing these
hypotheses and improving the accuracy of our results, for
example employing more realistic but comparably compact
bases (e.g. def2-svp split-valence polarized) combined with a
more flexible choice of η.." ← Results relying on artificial choice of basis sets and parameters means No quantum mechanical prediction
p.10-right-2nd-paragraph says -- Disagree with experiments
"using HQC methods and matching leading classical
WF methods (= No quantum computer advantage )" ← did Not say their "(inaccurate) hybrid-quantum results matching experimental atomic energy, because this research could Not get exact experimental atomic energy due to very rough approximation"
p.9-Table IX, p.10-Fig.6 -- Not compare with experiments
They compared
hybrid-quantum-SQD only with classical computer methods such as SCI, CCSD, DMRG (= more rough or inaccurate approximate version of the variational Full-CI or FCI method choosing free parameters with No quantum mechanical prediction, this-p.21 ) without comparing their (wrong) hybrid quantum results with experimental energy.
As a result, due to impractical quantum mechanics and its approximations that cannot predict anything, IBM hybrid quantum method called SQD could Not get nor predict exact atomic energy.
All researchers including IBM already know the impractical quantum mechanics can Not predict anything, and their error-prone quantum computers are hopeless forever (= so they are deceiving us ! ).
This is why they tend to use the misleading phrases such as " simulate (= Not predict ) atoms" and "discover drugs" in the overhyped fake news for investment and educational frauds which artificially- manufactured impractical mainstream science definitely has detrimental effect on real science curing diseases, education and people across the world.
Quantum computers based on fantasy parallel worlds are too error-prone to calculate anything (= even factoring 21 is impossible ), and useless forever ( this or this-1st, and 2nd-last-paragraphs ).
So any news on quantum computers calculating or simulating molecules or fusion is just overhyped fake news ↓.
This or this news on fake IBM quantum computing on fusion energy ↓
1st-paragraph says -- Fusion energy calculation ?
"IBM, and Cleveland Clinic are applying quantum computing to a critical challenge in fusion energy: efficiently sourcing its fuel."
3rd-paragraph says -- Classical computer needed
"The team.. fragmenting the calculation into smaller, manageable clusters solved by classical computers, with more complex clusters tackled by a quantum computer using sample-based quantum diagonalization (SQD = quantum computers and mechanics are useless ). The researchers successfully modeled nine configurations of the molten salt FLiBe, each a small cluster of 21 ions, and computed their energies with and without tritium"
4th-paragraph says -- Impractical research
"While simulating a full-scale, one-meter thick blanket containing trillions of particles remains beyond current computational capabilities ( this-10th-paragraph )"
↑ In this SQD method. the error-prone useless IBM quantum computers could only randomly sample numbers, while classical computers had to conduct all atomic (Hamiltonian) energy calculations ( this-12th-paragraph, this or this-Background ).
Quantum mechanics is useless, cannot solve its Schrodinger equation nor predict any atomic energy regardless of using quantum computers or not ( this-figure-middle~lower ).
↑ The quantum mechanical impractical approximate variational methods such as wrong Hartree-Fock and too time-consuming configuration interaction (= CI ) used in this IBM deceptive hybrid SQD method (= this-3rd-paragraph ) equal to QSCI (= Quantum Selected Configuration Interaction ) can only choose fake solutions or trial wavefunctions or unphysical Slater determinants out of infinite choices and infinite free parameters, which cannot predict anything including fusion energy ( this or this-p.4-1st-paragraph ).
↑ This IBM research paper ( this or this-2nd-paragraph-link ) ↓
p.6-3rd-paragraph says -- Quantum computer random sampling
"In SQD (= OSCI = deceptive hybrid method ), candidate electron configurations (= artificially-chosen fake trial wavefunctions or Slater determinants ) are drawn (= randomly sampled ) from a parameterized quantum state prepared on
a quantum processor (= useless ),"
p.6-3rd-paragraph says -- Classical computer needed
"a configuration recovery operation is used to mitigate errors, and a compact CI
Hamiltonian is then diagonalized classically. ← A classical computer must calculate fusion atomic energy from artificially-chosen fake trial wavefunctions called diagonalization (= this or this-lower-equation-56 ), which cannot predict any energies.
".. The quantum device thus acts as a configuration generator, while classical post-processing recovers the fragment ground-state energy... (= classical computers must calculate all atomic energies or Hamiltonian )"
p.6-4th-paragraph says -- Ansatz = chosen wavefunctions cannot predict
p.24-2nd-last-paragraph says -- Samples prepared by classical computers
"Parameter initialization is seeded from a classical CCSD calculation on the embedded cluster:... the LUCJ-state energy"
p.26-1st-paragraph says -- Classical computer calculated all
"The raw bitstring counts returned by the IBM
Sampler primitive are post-processed entirely on classical hardware to produce the extended sample-based
quantum diagonalization (ext-SQD) ground-state energy"
↑ So this deceptive hybrid method called SQD just choosing fake trial wavefunctions cannot predict any energies including fusion, and their useless quantum computers can only randomly sample or generate numbers with No meaningful computation.

Feel free to link to this site.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}