トップページ (2電子原子も含む正確な新ボーア模型)
多原子分子のページに戻る。
(Fig.1) 多原子分子。
下のサンプルプログラムを用いて、初期データを変えれば、様々な 多原子分子の計算、視覚化ができる。
( この方法は 後で述べる。)
最初に、メタン様、 H2O様、フォルムアルデヒド様分子について考える。
上のプログラムのソースコードを表示させれば、それをそのままセーブ、コンパイル可能である。
( コピー、ペーストも可能だが、少し長いと思われる。)
このクラス名は "orbit" なため、このテキストエディタを "orbit.java" とセーブしてコンパイルしてほしい。
もし、詳細は -Xlint:unchecked " オプションを指定して再コンパイルしてください。というメッセージが表示されても そのまま無視して実行できる。
(Fig.2) メタン分子 (= CH4 ) の選択。
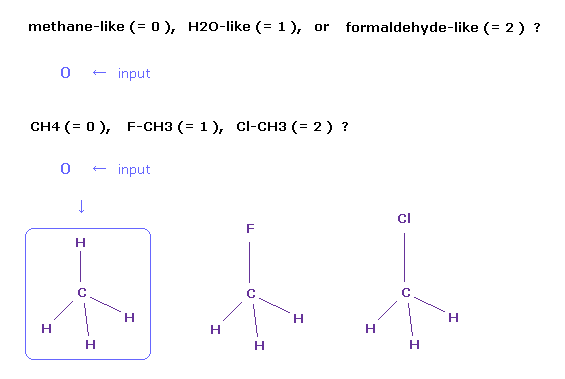
プログラム実行後、 " methane-like (= 0 ), H2O-like (= 1 ), formaldehyde-like (= 2 ) " という文章が 画面に表示される。
もし "0" を入力して エンターキーを押すと、 " CH4 (= 0 ), F-CH3 (= 1 ), Cl-CH3 (= 2 ) " という文が次に表示される。
ここで また "0" を入力して エンターキーを押すと、 メタン (= CH4 ) が 画面上に表示される。
他のメタン様分子は フルオロメタン (= F-CH3 )、クロロメタン (= Cl-CH3 ) があり、 "1" もしくは "2" を この時 入力すればいい。
(Fig.3) OFH (= 水素-フッ素-酸素 ) 分子の選択。
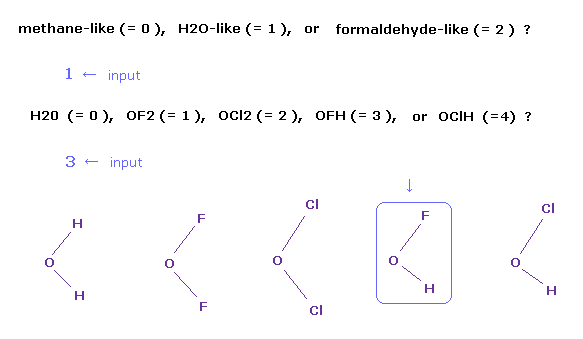
プログラム実行後、" methane-like (= 0 ), H2O-like (= 1 ), formaldehyde-like (= 2 ) " という分が 画面に表示される。
ここで "1" を入力して エンターキーを押すと、 H2O-様分子になる。
次に " H2O (= 0 ), OF2 (= 1 ), OCl2 (= 2 ), OFH (= 3 ), or OClH (= 4 ) ? " が表示される。
ここで "3" を入力して エンターキーを押すと、OFH 分子が 画面に表示される。
フォルムアルデヒド様分子 (= 2 ) の場合は、フォルムアルデヒド ( O=CH2 )、 O=CF2 もしくは O=CCl2 を選択できる。
(Fig.4) フルオロメタン (= F-CH3 ) 分子。
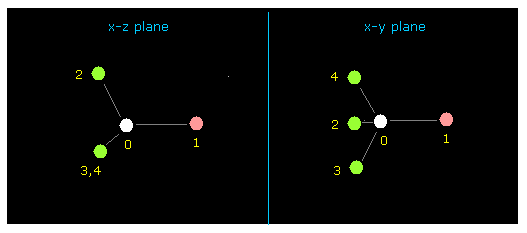
上記のプログラム実行後、 "0" (= メタン様 )、ついで "1" (= F-CH3 ) を入力する。
F-CH3 の断面図 ( x-z、x-y 画面 ) が表示される。
この画面は "分子の全体図" であり、 各原子の 絶対座標が テキストボックス ( +X, +Y, +Z ) 内に表示される。
( 単位: 1 MM = 1.0 × 10-14 meter )。
フッ素 (= atom 1 ) は ピンクの円、水素 (= atom 2, 3, 4 ) は緑、 炭素 (= atom 0 ) は 白の円で表示される。
(Fig.4') フルオロメタン (= F-CH3 ) 分子。
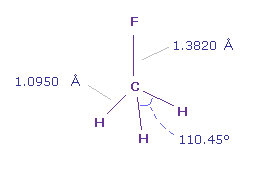
C-F 結合長は 1.3820 Å で C-H 結合長は 1.0950 Å である。
画面上には、 フッ素 (= F ) 原子が x 軸、 テキストボックス内の "nuc" は 各原子と 0 (= C ) 原子間の距離。
実際の H-F-H 角は 110.45°であるが、 ほぼ 正四面体の 109.45°に等しい。
そこで ここでは 通常の 109.45°で計算する。 ( もちろん、後で説明するが、これらの座標は変更できる。 )
これらの結合角 (°) は 最後の "angle" の行に表示される。
例えば、 "1-n-2" は atom 1 (= F ) - C nuc - atom 2 (= H ) の間の角度である。
(Fig.5) 力 = 1000.
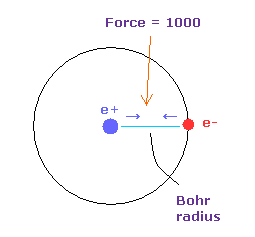
テキストボックス内の ( FX, FY, FZ ) は 各原子核に作用する力の成分を表している。
各原子が安定になるには これらの値は なるべくゼロに近づく必要がある。
力の単位は Fig.5 に示してある。
水素原子の 電子と原子核間に働く力を "1000" とする。
つまり 電子と +e の原子核間の距離が ボーア半径のとき、その力は 1000 になる。
例えば、1468 の力は H 原子の基底状態のものの 1.468倍という意味である。
"tV" は F-CH3 分子の全ポテンシャルエネルギーである。
また "V-1" は 2つの原子の分子 ( 例えば ここでは C と F = 1 ) 中のポテンシャルエネルギーを意味する。
この "分子の全体像" の画面は もっと大きい分子用の画面のため、小さい F-CH3 の分子の場合は この画面はあまり使用しない。
(Fig.6) 各原子核とその価電子。
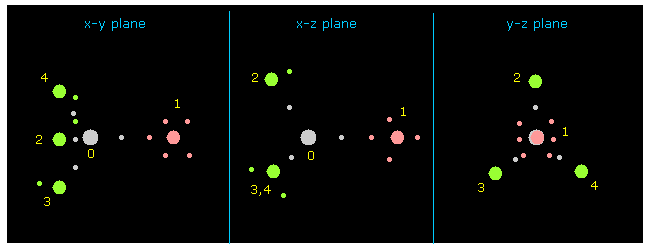
次に 右上の "nucleus"ボタンをクリックする。
"0" が "nucleus" ボタンの横に入力されているため、 0 atom (= C ) を中心に x-y、 x-z、 y-z 平面が表示される。
他の原子と その価電子 (= 小さい円 ) は C 原子核の周囲に表示される。
( FX, FY, FZ ) は 各原子核に作用する力の成分、 "cf" は C 原子核方向への力の成分である。
( もし この "cf" が 正の値なら、 その原子は C 原子核方向へ引きつけられている。 )
ある値を ( +X, +Y, +Z ) のテキストボックス内に入力して エンターキーを押すと、各原子核の相対座標を変更できる。
(Fig.7) Atom 1 を y 軸方向へ向ける。
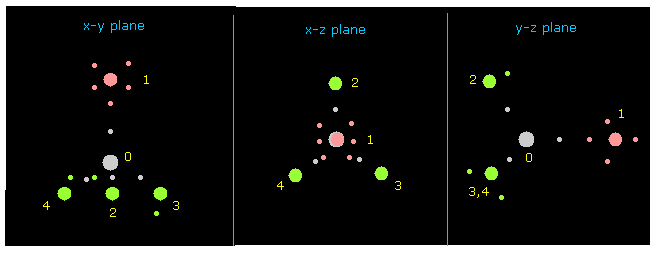
次に "1" を "direction" ボタンの横に入力して "direction" ボタンをクリックする。
このボタンは 分子全体を 回転させ、 指定した原子 ( この場合は atom 1 = F ) を y 軸方向へ向けさせる。
このボタンの他にも、"x-y ang"、 "x-z ang"、 "y-z ang" ボタンを入力すると 分子全体を 任意の平面上で 回転することができる。
ある角度 (= °) を入力して これらのボタンをクリックすればいい。
(Fig.8) フッ素の画面 (= atom 1 )。
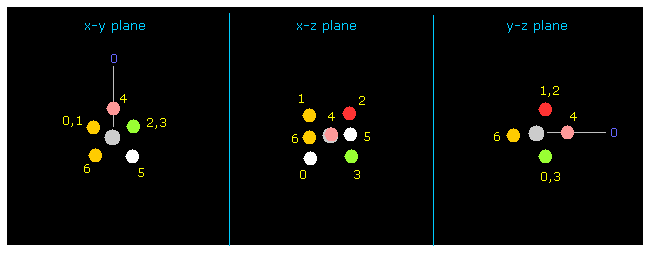
次に "1" の値を "electron" ボタンの横に入力して、 "electron" ボタンをクリックする。
そうすると、 atom 1 (= この場合はフッ素 ) と その 7 つの価電子が 画面上に表示される。
炭素 (= atom 0 ) は この電子画面でのみ y 方向へ向けられる。
"nucleus" もしくは "top nucl" ボタンをクリックすると、 Fig.7 (= 各原子核画面 ) もしくは
最初のセクションの 分子全体像へ再び戻れる。
任意の値 (MM) を テキストボックス (+X, +Y, +Z) 内に入力して エンターキーを入力すると、各相対座標を変更できる。
他のパラメーターの意味は このページのと同じである。
(Fig.9) フッ素を x-y 平面で 90°回転させる。
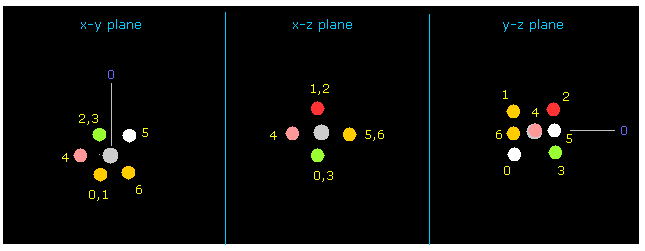
Fig.7 と Fig.8 に示したように、 最初の画面では フッ素の価電子 "4" が 炭素の方向へ向いている。
そのため この電子 "4" と 炭素の価電子間の 反発力が強くなり、分子を不安定にさせる。
90°を "x-y ang" ボタンの横に入力して "x-y ang" ボタンをクリックする。
すると フッ素は x-y 平面で 90°回転し、フッ素と炭素の価電子は 互いに避け合うことができる (= Fig.9, 10 )。
(Fig.10) フッ素を x-y 平面で 90°回転させ 反発力を回避する。
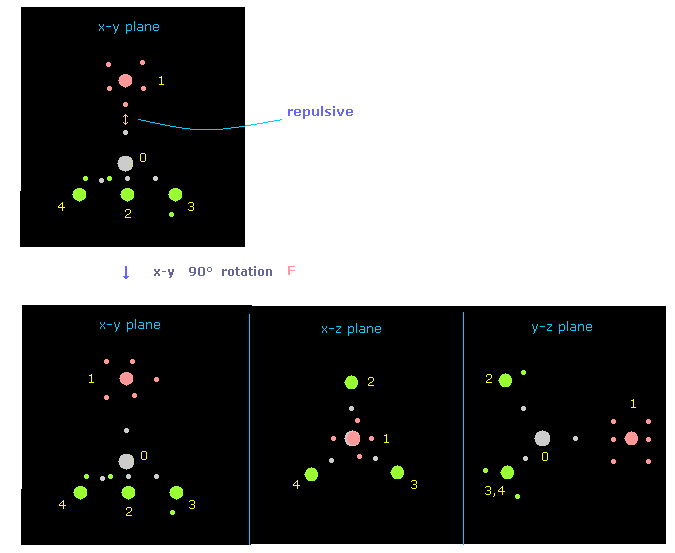
"nucleus" ボタンの横に "0" を入力して "nucleus" ボタンをクリックすると、炭素と その4つの隣りあった原子の視点から フッ素のみが x-y 平面で 90°回転したことが分かる。
( 注意: 正の角度は 電子画面で 反時計方向である。 そのため このケースでは、原子画面では 逆方向に回転している。)
"nucleus" ボタンは 指定の原子 (= その原子番号を指定のテキストボックス内に入力 ) と その 周囲の原子を表示させる。
また "top nucl" ボタンは 指定した原子を中心として 分子全体を表示させる。
( CH4 や F-CH4 などの小さい分子では、 これらの "nucleus" と "top nucl" ボタンは ほぼ同じ画面になる。 )
(Fig.11) C 原子核に作用する力は "釣り合って"いる。
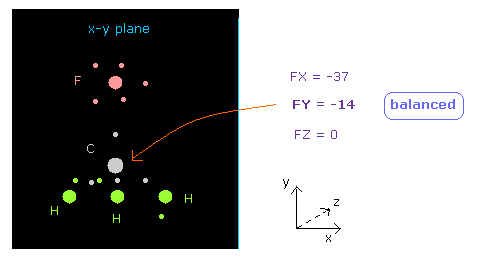
原子画面で 下の "now 0" の行を見るように。これは atom 0 (= 炭素 ) が中心にあるという意味である。
"now 0" の隣りのテキストボックス内に この炭素に作用する力が表示される。
FY = -14 は、ほぼ ゼロの値である。
つまり このケースでは F と 3つの H の間で 中心の炭素原子核は ちょうど 釣り合っていることを意味している。
"12000" の値を "atom 1" の "+Y (MM)" のテキストボックスに入力して" エンターキーを押すと、 F-C の結合長が 12000 MM (= 1.2000 Å ) に変わる。
このケースでは 炭素に作用する力が F 原子核の反発力のために 大きくなってしまう ( FY = -125 )。
つまり 結合長 13820 MM (= 1.3820 Å ) が 釣り合った長さである。
(Fig.12) 4つの価電子に作用する力が "釣り合って"いる。
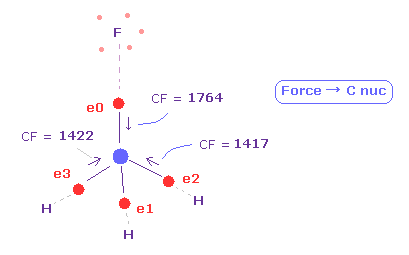
再び、 "13820" を "atom 1" の "+Y (MM)" のテキストボックス内に入力して、エンターキーを押す。
次に "0" を "electron" ボタンの横に入力して "electron" ボタンをクリックする。
すると 炭素原子の 4つの価電子 ( 0-3 ) が表示される。
( その周囲の 青の数字は その周囲の原子番号を意味している。 )
各電子の 力 "CF" は 各価電子に作用する力の C 原子核方向への成分である。
Fig.12 に示したように、 F-C と H-C 結合長が 1.3820 Å と 1.0950 Å のとき、これらの力は ほぼ 互いに 等しくなる。
F 側では CF = 1764、 H 側では CF = 1417 である。
(Fig.13) 4つの価電子に作用する力のバランスが壊れる。
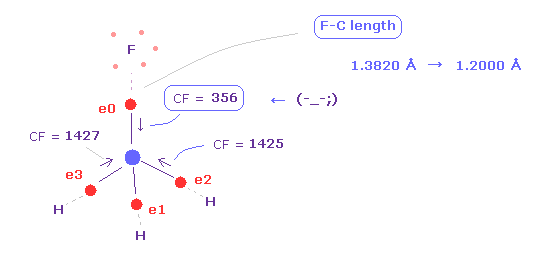
再び "nucleus" ボタンをクリックして 原子画面に戻る。 そして +Y 座標を "13820" から "12000" にする。
これはつまり F-C 結合長が 短くなった ( 1.3820 Å → 1.2000 Å ) ことを意味している。
そして "electron" ボタンをクリック ( その隣のテキストは "0" で ) して、 炭素の価電子に作用する CF を 再び表示させる。
Fig.13 に見るように、F-C 結合長が短くなると (= 1.2000 Å )、炭素の価電子 "0" は フッ素の原子核に 引きつけられすぎることになる。
そのため CF = 356 は 他の CF = 1425 に比べて 弱くなってしまう。
( もちろん、フッ素原子核も 電子側に 強く引きつけられすぎることになる。)
これはつまり これらの粒子が不安定になったことを意味している。
このページに示したように、これら価電子と原子核の不安定性は C-H = 約 1.0900 Å などの 結合長を決定しているといっていい。
実際に C-H 結合長を 1.0950 Å から 0.9500 Å に短くすると、この H 側の 炭素の価電子の CF が 負 = -118 になってしまう。
( この奇妙なケースでは、炭素の価電子が 炭素ではなく 水素の原子核に引きつけられてしまう。 )
上で述べたように、3つのパターンの画面がある。
"nucleus" ボタン (= 各原子核と その周囲の核 )、 "electron" ボタン (= 各原子と その周囲の価電子 )、もしくは "top nucl" ボタン (= 分子全体像 ) の3つである。
(Fig.14) 電子画面 (= F )。
"electron" の画面では、 "nuc" は 各電子と その原子核間の距離を表している。
指定のテキストボックス内に ある値を入力して この "nuc" ボタンクリックすると この nuc の値を変更できる。
中心電荷 Z も ある値を入力して "charge Z" ボタンをクリックすれば 変更できる。
"inversion" ボタンをクリックすると、 画面に表示されている 電子が その核に対して 反対方向に移動する。
スクロールバーの中から 原子の種類を選んで "A atom" ボタンをクリックすると、原子の種類を変更できる ( H, C, N, O, F, Si, P, S, Cl )。
これらの価電子間の位置関係を変更したいときは、"x-y ang"、 "x-z ang"、 "y-z ang" ボタンを用いて、適切な位置に それらを回転させるといい。
もしくは テキストボックス内の 相対座標を自由に変えることができる。
(Fig.15) 水素原子のタイプ、 0、1、2、3。
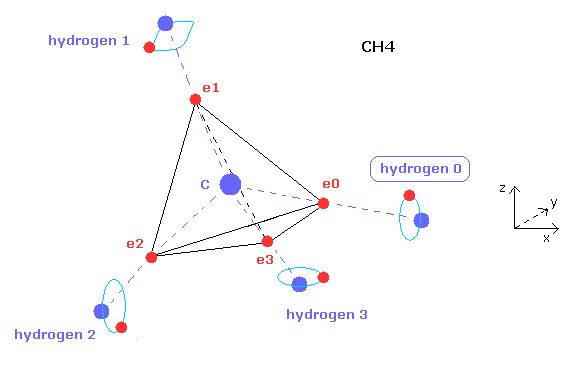
"electron" ボタンをクリックして、水素原子のどれかの電子を表示させて、0, 1, 2, 3 のどれかの数値を 指定のテキストボックス内に入力して "hydrogen" ボタンをクリックすると、水素原子の タイプ を変更することができる。
水素原子は 1つの電子しかないため、それらの対称的な位置を調整する必要がある。
例えば、メタン ( CH4 ) では、atom 1 が hydrogen 0、 atom 2,3,4 が それぞれ hydrogen 1,2,3 である。
hydrogen 0 の電子の座標のみ変更することができ、この hydrogen 0 の電子座標を元に hydrogen 1, 2, 3 の電子が 自動的に 対称的な位置に配置される。
Fig.15 の位置関係を元に、この水素のタイプを決めるように。
例えば、フルオロメタンでは、atom 1 は フッ素、 atom 2,3,4 は それぞれ hydrogen 0、1、3 である。
電子画面では ドブロイ波の 1軌道の数が "Waves" の列に表示される。
水素原子以外の多電子原子では、 "waves" は その原子内でのみの環境で計算される。
( 例えば、炭素では、周囲の原子からの力は ほぼ釣り合っているため、"Waves" は 炭素原子内でのみで計算する必要がある。)
また "direw" は 各電子に作用する 直接の力から計算した ドブロイ波の数である。
水素原子では、 "direw" と "Waves" は 同じである。
"dia" と "ave" は すべての価電子の "direw" と "Waves" のドブロイ波の平均値である。
水素原子では、それに作用する力で動いた後の電子で 計算した結果が 2行目である。
これらの方法は このページのものとまったく同じである。
(Fig.16) 多原子分子。
このプログラムも 表示したテキストを そのままセーブ、コンパイルできる。
( このテキストを "orbitt.java" とセーブして コンパイルしてほしい。 )
"--- You can change freely initial data -----" の分の内側のパラメーターを自由に変更できる。
(Fig.17) 多原子分子。

最初に 全原子数を "maxa" に入力する。
例えば、 O=CF2 は "4"、H2O は "3" である。
そして この maxa と同数の配列を用意する。
( 0, 1, 2 ) の成分は 絶対座標 (= x, y, z ) である。
"3" 成分目は 中心の正電荷 ( H=1, C=4.22, N=5.25, O=6.273, F=7.3, Si=5.05, P=6.12, S=7.18, Cl=8.26 )。
"4" 成分目は 電子の半径である。 単位は MM ( H = 4500, C=6415, O=4596, N=5332, F=4003, Si=11500, S=8622, P=9825, Cl=7640 )。
このページも参照のこと。
model2 では、 "0" 成分目は 価電子の数 ( H=1, C=4, O=6, F=7 ...)。
"1-6" 成分目は 隣接した原子の番号。
この場合は 炭素 (= atom 0 ) の 隣は 酸素 (= atom 1)、 2つの F (= atom 2, 3 ) である。
そして 他のすべての "1-7" の成分に "-1" を入力する。
最後の "8" 成分目は 水素原子のタイプ 0-3 である。
例えば、 CH4 では atom 1-4 が それぞれ hydrogen 0-3 であり、hydrognen 0 の電子座標のみ変更でき、他の水素の電子は それを元に 自動的に対称的な位置に配置される。
ある原子番号を入力して "top nul" ボタンをクリックすると、その指定の原子を中心にして 分子全体が表示される。
ある数値 (MM) を "move" ボタンの隣の テキストボックス ( 左から x、 y、 z ) 内に入力して"move" ボタンをクリックすると、分子全体を移動することができる。
"overall" ボタンをクリックすると、 "overall" と "each at" の変更ができる。
"each at" 状態では、"each at" の下のテキストボックスに ある原子番号を入力して "move" ボタンをクリックすると、その指定の原子のみ移動できる。

2013/5/12 updated This site is link free.