
Home page
Quantum computer is useless
Quantum phase estimation is useless
(Fig.1) The recent overhyped research "Hybrid quantum computing for drug discovery" is fake, using only 2 useless qubits and impractical quantum mechanics that cannot predict any molecules nor drugs.
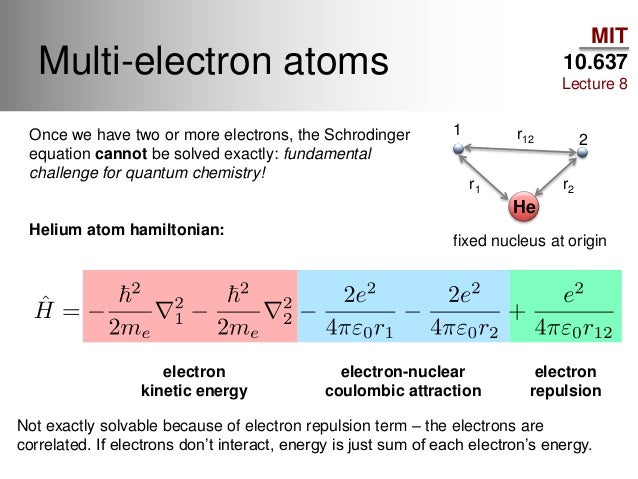
Unrealistic quantum mechanics relies on solving ancient Schrödinger equation whose total energy (= Hamiltonian H ) consists of kinetic energy (= space derivative based on de Broglie wave theory ) and Coulomb potential energy between electrons and nuclei to predict atomic energy, which is impossible.
Except for one-electron hydrogen atom (= agreeing with real Bohr's model, this-p.12-last ), quantum mechanics cannot solve Schrödinger equations for any multi-electron atoms such as helium or molecules, nor predict energy ( this-p.21, this-4th-paragraph, this-p.26-2.5 ).
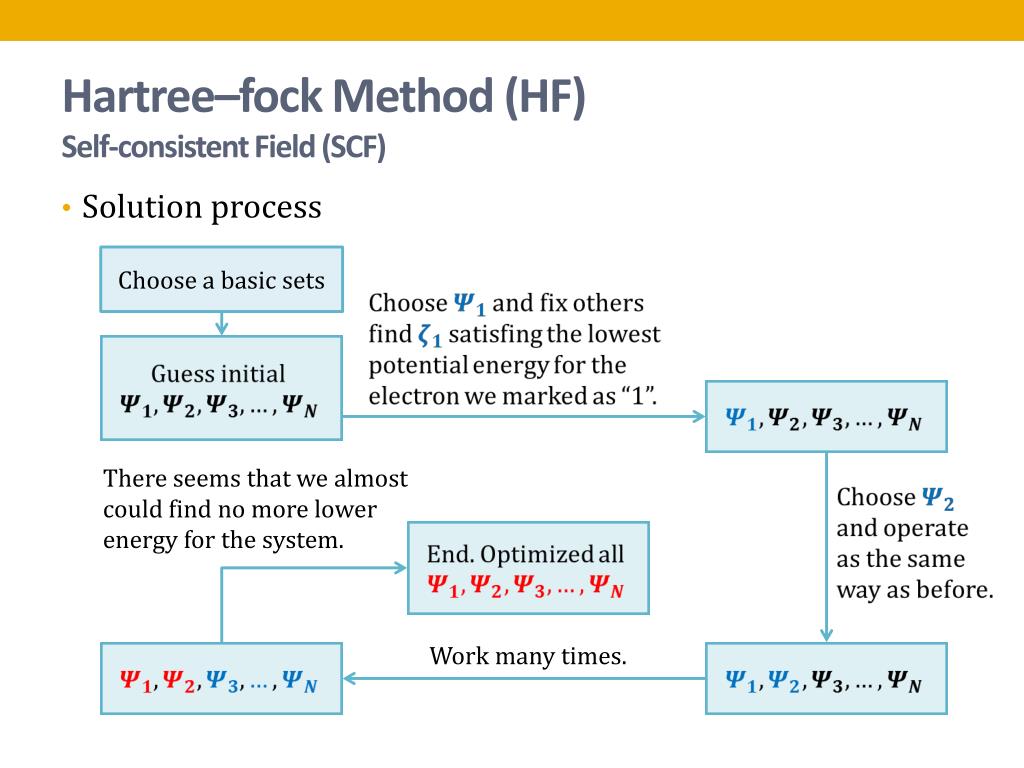
Quantum mechanics can only artificially choose fake trial (= basis set ) wavefunctions for unsolvable Schrödinger equation out of infinite candidates and infinite free parameters ( this-4~5th-paragraphs ) to find ones giving the lowest (fake) total energy E in its approximate variational method ( this-p.4-4th-paragraph ), which takes infinite time, impractical, cannot predict any atomic or molecular energy ( this-p.9-last, this-p.22-upper, this-p.6-4.4.1 ), proven wrong.
↑ These useless approximate quantum mechanical methods for unsolvable Schrödinger equations are called variational method (= proven wrong ), Hartree-Fock, configuration interaction (= CI ), all of which are impractical ( this-p.2-introduction ).
In these quantum mechanical approximate methods, the chosen wavefunctions (= expressed as unphysical antisymmetric or Slater determinants ) giving the lowest energy must consist of infinite terms and infinite adjustable parameters, which are impractical ( this-p.6-4th-paragraph, this-1.4, this-p.6-2.2~p.7 ), and can never find true wavefunctions nor predict anything.
This impractical mainstream quantum mechanical approximate variational method choosing arbitrary fake trial (basis set) wavefunctions ( this-p.4~p.6 ) for unsolvable Schrodinger equation cannot predict true atomic or molecule energy, so meaningless, just a waste of time ( this-(9.80), this-last-sampling vs variational inference ).
This-p.2~p.3 say -- No quantum mechanical prediction
"we cannot solve the problem
exactly (= Schrodinger equations are unsolvable )"
"The variational principle... Problem: you do NOT know how
close your result is compared to the exact result"
This or this-p.4-1st-paragraph says -- Useless variational method
"So we do not have any way of
knowing how good or how bad our trial wave function is.
The variational method also does not tell us what kind of a trial function we must
choose ( this-p.3-lower )"
This-1.Variational principle-last says -- Choose fake wavefunction
"Its accuracy heavily depends on the choice of the trial wave function
Ψtrial. A poorly-chosen trial function may lead to an energy estimate that is far from accurate."
This or this-p.7-last-5 caveat says -- Variational method cannot predict
"One word of caution about the variational method is that there is No way
to judge how close your result is to the true result."
So quantum mechanics can never predict any molecular energy nor discover effective drugs, whether (fictional) quantum computers or classical computers are used ( this-p.1-right~p.2 ).
Today's quantum computers are error-prone, useless, cannot even factor the simplest 21, much less discover effective drugs or calculate molecular energy, contrary to an incredible amount of overhyped fake news.
This-lower challenges (2026) say -- Useless quantum computer
"Hardware limits: Today’s qubits are noisy and error- prone"
Today's quantum computers with only less than 200 qubits (= one qubit can take only 0 or 1 value ) are still Not computers, and far from millions of qubits required for a practical quantum computer which is impossible to realize forever ( this-8th-last-paragraph ).
This or this-lower-Challenges (2026) say -- No quantum advantage
"they (= quantum computers ) have yet to demonstrate a practical advantage over classical methods"
"Error rates, scalability,.. pose ongoing challenges."
This recent news (2026) on hopeless quantum computers says ↓
3th-paragraph says -- Impractical quantum computer
"the most popular quantum algorithms face serious obstacles when trying to compute the lowest energy state of molecules"
8th-paragraph says -- Useless quantum VQE method
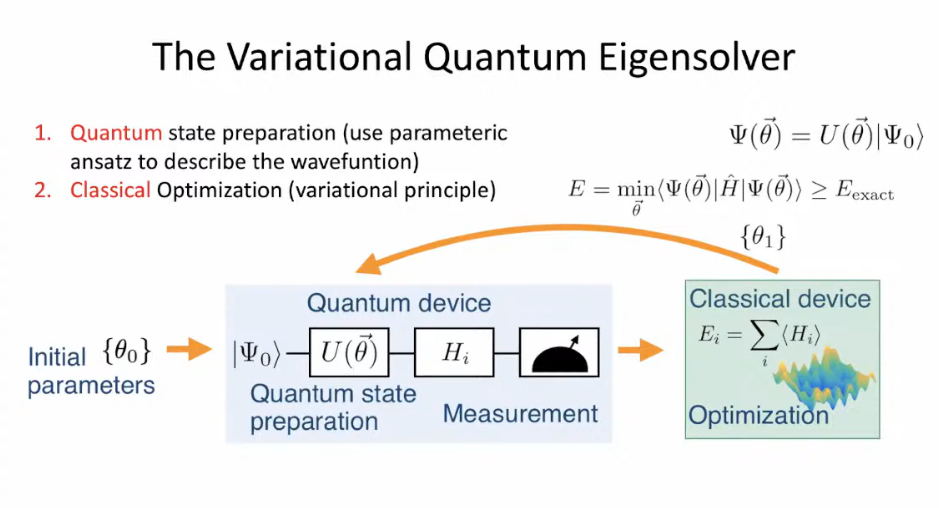
"The first method, VQE (= hybrid quantum molecular Hamiltonian energy calculation method called variational quantum eigensolver based on impractical quantum mechanical variational method just choosing fake trial wavefunctions for unsolvable Schrodinger equation, this-4th-paragraph ), is designed for near-term quantum computers that are still noisy and prone to errors" ← Useless
9th-paragraph says -- Classical computer needed
"VQE works through a hybrid approach (= almost classical computer method ) where a quantum computer prepares a candidate quantum state for the molecule, while a classical computer adjusts parameters step by step to minimize the calculated energy. The idea is to gradually approach the molecule’s true ground state."
11th-paragraph says -- Too time-consuming quantum
"For instance, when the researchers examined the chromium dimer molecule (Cr₂), they found that.. When the many required optimization steps are included, the total runtime could stretch to around 24 years ( this-p.3-left-1st-paragraph )"
13th-paragraph says -- Inaccurate quantum VQE method
"However, the study shows that VQE frequently struggles to handle them accurately (= so hybrid quantum VQE is impractical and unable to predict any energy, contrary to overhyped news, this-last-paragraph )"
The recent overhyped research "A hybrid quantum computing pipeline for real world drug discovery ( this-11th-paragraph )" is useless, far from real drug discovery.
And this research used a fake quantum computer with only 2 qubits (= one qubit can take only 0 or 1 value, so still Not a quantum computer, this-p.4-Figure 3 ), which has No ability to calculate any molecular energies nor discover drugs, contrary to the overhyped headline.
In this recent overhyped research on hybrid quantum computer for drug discovery, they tried to calculate energy of some small drug-related molecules by the impractical quantum mechanical variational approximate method ( this-p.2-1st-paragraph ) called variational quantum eigensolver (= VQE ).
They chose just 2 fake trial wavefunctions (= ψ1, ψ2 ) for only 2 electrons' orbitals for just 2 qubits (= Not a quantum computer ! ) as active space and fake effective (core) potential Veff for roughly expressing other electrons in a small prodrug molecule for unsolvable Schrödinger equations that cannot predict any molecular energies ( this-p.3-2nd-last-paragraph,p.10 ).
And they calculated (fake) molecular total energy E by quantum Hartree-Fock approximate method integrating the chosen fake wavefunctions and Schrodinger energy equation (= H, h ) by an ordinary classical computer ( this-p.6-lower~p.7, this-p.4-(2) ).
Their quantum computer with only 2 qubits (= 01, still Not a computer ) can Not calculate molecular energy nor discover drugs in this research.
In this recent research on deceptive hybrid quantum computing pipeline for drug discovery, their quantum computer had only 2 useless qubits (= one qubit can take only 0 or 1 value, which is still Not a computer ) that cannot calculate molecular energy nor discover drugs, contrary to the overhyped news.
↑ Just 2 qubits cannot calculate any molecular energy, so in this hybrid method called variational quantum eigensolver (= VQE, this-4th-paragraph ), all molecular energy calculations and optimizing parameters of chosen (fake) trial wavefunctions must be conducted by an ordinary classical computer ( this-The VQE algorithm, this-middle-hybrid approach ), which cannot predict anything ( this-p.1-abstract ).
In this hybrid variaitonal quantum eigensolver (= VQE ) for the overhyped drug discovery, they had to artificially create fictional approximate quantum computer-version-Hamiltonian energy usable for just 2 qubits (= a, this-middle ), which contains energy parameters (= h ) calculated by a classical computer integrating the chosen fake wavefunctions with unsolvable Schrodinger energy equation in ordinary impractical quantum mechanical Hartree-Fock approximate method ( this-lower, this-Hamiltonian-mapping, this-p.9-p.10, this-p.11 ).
This-p.6-lower~p.7 says -- Classical computer needed
"VQE relies on quantum hardware.. The Hamiltonian (= total energy ) is mapped onto
a qubit (= only 2 qubits ) representation using transformations like Jordan-Wigner mapping. The energy
is evaluated as"
"The one and two electron integrals, hpq and hpqrs (= impractical quantum Hartree-Fock energy integral ) respectively, are
easily calculated on classical computers.. through
solving the Hartree-Fock equations"
This-p.3-right-3rd-paragraph says -- Energy calculated classically
"N is the number of qubits and also the number of molecular basis wavefunctions considered. The hpq and hpqrs terms
can be computed classically (= using classical computers instead of the useless quantum computer ) via electron integral formulas ( this-p.13-(88), this-p.4-(2) )."
Another deceptive hybrid quantum computer method called quantum phase estimation (= QPE ) also has to calculate all Hamiltonian energy parameters (= h ) by a classical computer (= based on artificially-chosen fake trial wavefunctions that cannot predict any energy ) instead of today's error-prone useless quantum computers ( this-p.1-right-last,footnote, this-p.3-right-2nd-paragraph ).
↑ This useless QPE can only change qubits by a phase eiHt (= this Hamiltonian energy H must be artificially created by a classical computer ) and measure them, which cannot predict any molecular energy ( this-p.7-2nd~3rd-paragraphs ).
In this (deceptive) hybrid quantum computing variational quantum eigensolver (= VQE ) they expressed the fictitious Hamiltonian (= total ) energy (= created by a classical computer ) by some free parameters (= θ ), and tries to optimize this free parameters θ by a classical computer's (quantum) variational methods to get the lowest total energy E ( this-lower, this-p.1-p.2, this-p.3-Fig.2 ).
↑ This overhyped drug-discovery quantum computing with only 2 qubits in hybrid VQE method contributed nothing to molecular energy calculation, let alone drug discovery.
They just changed 2 qubits' phases based on the free parameters θ optimized by a classical computer ( this-p.9-Methods ) using the impractical quantum variational method that cannot predict any energy, and measured these 2 qubits, which was supposed to express the total energy E in the qubit-version fake simple Hamiltonian equation.
Quantum mechanics, whose Schrodinger equations are unsolvable, heavily relies on choice of fake trial wavefunctions that cannot predict any molecular energies in its approximate variational method or variational quantum eigensolver.
Actually even this hyped news-lower Not ready quite yet admits
"using quantum computers for current drug discovery faces significant limitations. For example, quantum computing is still plagued by longer computational times and errors, which impede its accuracy and efficiency in drug discovery."
↑ This hyped research paper (= this-last link ) on quantum computing drug discovery ↓
p.1-last~p.2-upper says -- Classical computer needed
"Our hybrid quantum computing pipeline.. , the core of
VQE is to employ parameterized quantum circuits to measure the energy of the target molecular system. Then, a classical optimizer is employed to minimize the energy expectation until convergence. Due to the (impractical) variational
principle.. (= a classical computer is needed to get the lowest ground-state energy by quantum variational method that cannot predict any energy)"
p.3-2nd-last-paragraph-upper says -- Inaccurate quantum computer
"Despite that quantum devices with more than 100 qubits are becoming available, simulating large chemical
systems would require very deep circuits, which will inevitably lead to inaccurate outcomes due to intrinsic
quantum noise (= today's error-prone useless quantum computers )"
p.3-2nd-last-paragraph-middle says -- Choose fake orbits, No prediction
"we employed the active space approximation due to its popularity and versatility, which simplifies the
QM region into a more manageable two electron/two orbital system (= just choosing 2 fake electrons' orbits, Not a drug molecule ).. The CASCI energy can be considered as the
exact solution under the active space approximation (= CASCI approximate method artificially choosing only some 2 electron fake orbital wavefunctions in active space for impractical quantum CI calculations, which artificial choice can not predict any molecular energy, this-p.2-last-paragraph, this-3rd-paragraph )"
p.3-2nd-last-paragraph-lower says -- Only 2 useless qubits
"The wave function of the active space can then be represented by a 2-qubit superconducting quantum device (= just 2 qubits is still Not a quantum computer )."
p.3-last-paragraph says -- No quantum prediction
"For both classical and quantum computations, we selected the 6-311G(d,p) basis set (= artificially selection of fake trial wavefunctions with No quantum mechanical prediction )"
"Additionally, we included the results from HF and CASCI (= artificially choosing wavefunctions that can not predict anything, this-p.16.p.33 ), which are based on classical computational chemistry, for comparison"
p.4-Figure.3 shows -- Only 2 qubits = Not a quantum computer
This research used a quantum computer with only 2 qubits (= one qubit can take only 0 or 1 value ), which is still Not a quantum computer nor able to calculate anything.
p.5-3rd-paragraph-last says -- Slower quantum computer
"Nevertheless, for all
molecules, quantum computation takes approximately one minute longer than classical computers (= Just 2 qubits were more time-consuming than a classical computer )"
p.5-4th-paragraph says -- Impractical quantum computer
"In this study, we have limited the utilization of quantum computers to a few qubits employing the active space
approximation (= so cannot predict anything ), due to the limited size and gate noise of currently available quantum computers. "
p.5-last ~ p.6-top says -- Just 2 qubits, No prediction
"We first run the QM/
MM (= classical molecular mechanics based on artificially-chosen force field potential fitted to experiments that cannot predict anything, this-report-5th-paragraph, this-p.30-31, this-p.5-p.6 ) simulation on classical computers to get the baseline statistics, then move the QM energy computation to quantum computers... and the active
space wavefunction is processed using 2 qubits (= Just 2 qubits is still Not a quantum computer nor able to simulate molecules or drugs )."
p.8-Table 1 shows -- Inconsistent results.
Different methods gave different energies, which is Not prediction.
DFT is today's mainstream quantum mechanical approximation treating the whole molecules as one fake electron model with artificially-chosen fake exchange-correlation energy potential (= which cannot predict any energy ) lacking real atomic shape.
p.9-Discussions-3rd-paragraph says -- Inaccurate quantum computer
"However, it is important to note that the accuracy of VQE calculations
and the resources consumed require further improvement." ← hybrid quantum VQE caused erroneous values due to qubits' errors.
p.9-last-paragraph says -- Classical computer needed
"The VQE algorithm uses a parameterized quantum circuit.. The parameters of the quantum circuit θ are optimized to its optimal
value θ
using a classical optimization (= classical computer was needed to optimize variational energy parameters θ due to the useless 2-qubit quantum computer ) algorithm,.. to minimize
the energy.. According to the Rayleigh-Ritz variational principle (= impractical, this-p.3, which cannot predict any energy )"
p.10-2nd-paragraph says -- Fake useless 2-qubit Hamiltonian
"The
frst step is to calculate the integrals in the Hamiltonian under the molecular orbital basis (= by a classical computer this-p.4 ). Then, the second quantized fermion Hamiltonian is mapped to a spin Hamiltonian using fermion-qubit mapping (= creating fake Hamiltonian energy for 2 qubits with the help of a classical computer, because 2-qubits alone cannot calculate any molecular energies )"
p.10-4th-paragraph says -- Fake potential, No prediction
"this
approximation is sometimes also called the frozen core approximation... provides an effective repulsion potential Veff (= this approximate pseudo-potential energy often causes errors, this-p.1-right-1st-paragraph, this-p.7, this-2nd-paragraph )" ← Creating fake approximate potential cannot predict energy.
p.12-Classical computation details -- One-fake-electron DFT, useless
"we employed the B3LYP functional within DFT, chose the 6-31+G(d)
basis set for the molecular orbitals.. Grimme’s
D3 dispersion correction" ← Today's most-widely used quantum mechanical one-fake-electron DFT approximation with empirically-chosen fake exchange-correlation energy functionals which cannot predict any values.
↑ As a result, this overhyped hybrid quantum computing for drug discovery used only 2 useless qubits (= still Not a quantum computer ) and impractical quantum mechanical variational method artificially choosing fake trial wavefunctions whose energy was calculated by a classical computer, which can not predict any molecular energy nor discover drugs.
Quantum mechanics cannot solve its Schrodinger equation nor predict any multi-electron atomic or molecular energies.
The quantum mechanical approximation called variational method is impractical, taking too much time to choose fake trial wavefunctions and find ones giving the lowest energy out of infinite candidates and infinite free parameters, which also cannot predict anything.
So it is far better to use experimental values, energies, properties, and atomic shape from the beginning than to waste too much time in meaningless impractical quantum mechanical calculations.
Quantum mechanics has to artificially choose unphysical antisymmetric wavefunctions or Slater determinants for unsolvable Schrodinger equations, where each electron must unrealistically exist in all different atoms, which forbids each atom from having its shape or boundary.
Due to the impractical time-consuming Schrodinger equation, quantum mechanics has to treat the whole molecules as one-fake-electron DFT or unreal quasiparticle model lacking real atomic shape.
↑ This current mainstream quantum mechanical unreal atomic model hampers developing useful multi-probe atomic force microscopes (= deadend now ) that need real simple atomic model treating each molecules as a real object with shape.
We should immediately discard this impractical unreal quantum mechanical shapeless atomic model, and use real atomic model with shape (= based on experiments using practical multi-probe atomic force microscopes ) to develop nanotechnology and cure diseases instead of wasting too much time and money in the hopeless deadend quantum computers and information.

Feel free to link to this site.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}