(Fig.1) Today's experiments or AI cannot clarify atomic mechanisms of diseases.
The 1st, 2nd, 5th, 10th, 12-13th paragraphs of this hyped news (10/24/2025) say
"While symptoms can now be better managed with lifelong treatment, there is No cure to fully eliminate the (HIV) virus from the body"
"One of the most promising treatment avenues is disrupting HIV replication by impairing the function of integrase, a protein named for its role in integrating viral genetic material into the human host genome" ← false, these integrase inhibitor HIV drugs already exist, and cannot eliminate HIV viruses from bodies.
"Determining how integrase interacts with RNA will (= just speculation ) help us better understand this new role and inform the design of novel and more effective HIV therapeutics." ← hype
"Very little is known about what integrase is doing in the later stages of HIV replication" ← HIV proteins' functions are still unknown in today's deadend medicine, which is why there is still No cure for HIV.
"The researchers used cryo-electron microscopy (= cryo-EM is useless ) to collect two distinct integrase structures:" ← Science stops here
"First, they determined integrase's architecture as it forms the "intasome"—a special assembly of proteins and viral DNA."
↑ This research paper ↓
p.3-left-last~right says --
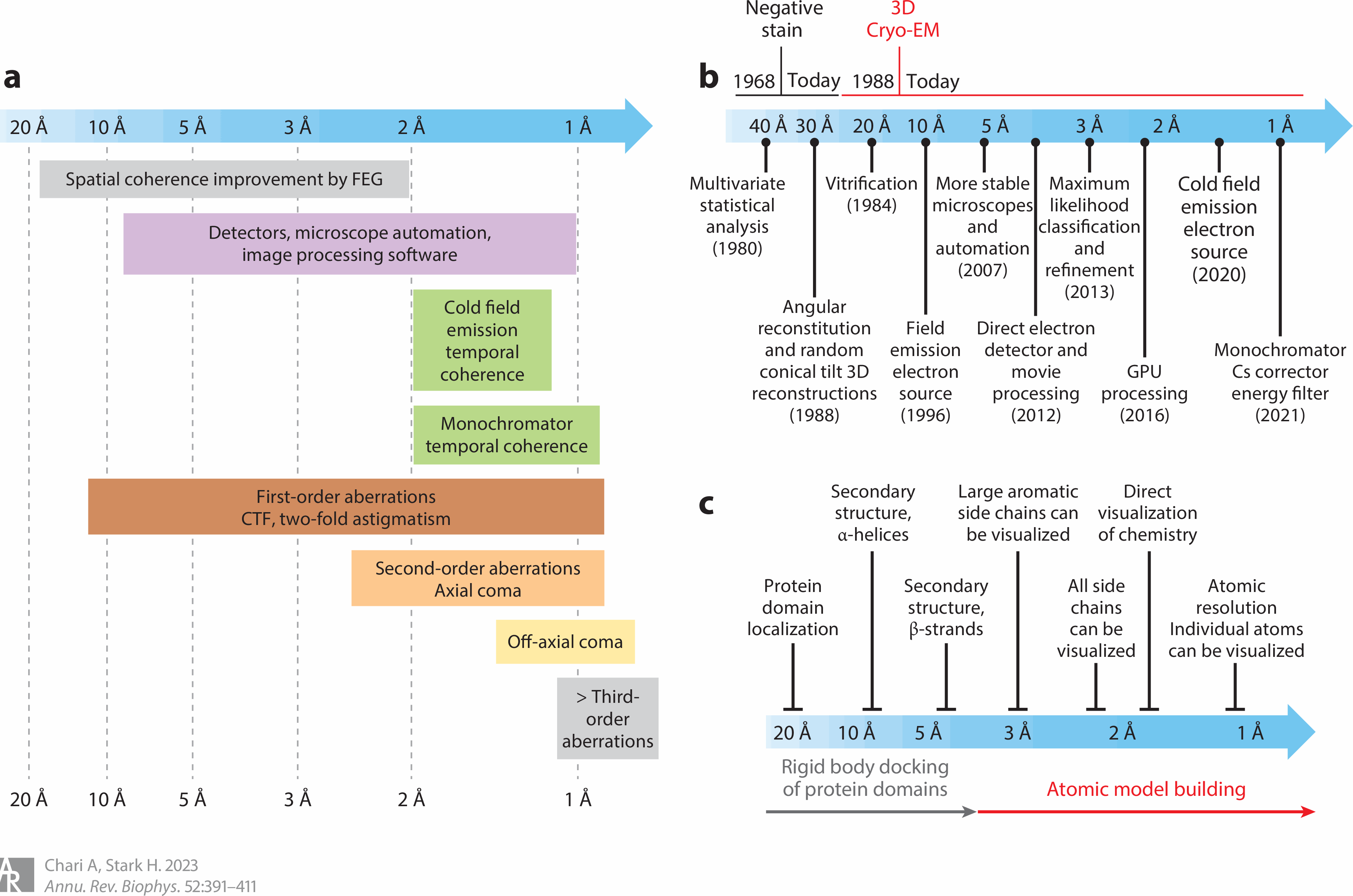
Bad-resolution in cryo-microscope
"We also collected a large cryo-EM dataset of apo IN in the absence of bound IBD (= in-binding domain ) and reconstructed a map to ~7 Å resolution (= too bad resolution to clarify atomic mechanism that needs 1Å resolution )"
p.10-right-study limitations say --
No HIV drug in this research
"ability to inform the pathway of intasome assembly from the
tetramer structure is limited" ← still HIV mechanisms are unknown, and No drug design at all in this research, contrary to the above hyped news.
Today's experiments relying on cryo-electron microscopes cannot clarify atomic mechanism that needs less than 1Å resolution.
↑ Even this latest HIV research got only vague protein images of very bad resolution of 8 ~ 12Å (= far worse and bigger than a single atom of 1Å, this-p.16-Fig.15d-local resolution of proteins by cryo-EM is 8~12Å = too bad, cannot see atoms )
The 7-8th paragraphs of this news (10/29/2025) say -- AI prediction is wrong
"The researchers modified the amino acid sequence of hundreds of sample proteins in such a way that the binding sites for their ligands exhibited a completely different charge distribution or were even blocked entirely. Nevertheless, the AI models predicted the same complex structure— as if binding were still possible." ← AI prediction is wrong.
"This shows us that even the most advanced AI models do Not really understand why a drug binds to a protein; they only recognize patterns that they have seen before"
↑ So today's AI or Alphafold trained on experimentally-obtained protein structures (= with too bad resolution, so useless ) can Not predict new protein structures nor develop effective drugs for mutated cancer cells.
↑ Because today's basic physics = (fantasy) quantum mechanics, unreal quasiparticle, DFT model is completely useless for expressing real molecular structures, hence, hampering developing useful multi-probe atomic force microscopes clarifying detailed atomic structures.
↑ Today's deadend physics and medicine is why researchers focus only on hopeless parallel-world quantum computers in vain.

Feel free to link to this site.
{kind=link}