
トップページ (2電子原子も含む正確な新ボーア模型)
タンパク質相互作用ページに戻る。
(Fig.1) 2つのタンパク質相互作用面の状態とは ?
ここでは タンパク質構造を調べるのに 次のサンプル JAVA プログラムを使用する。
サンプル JAVA プログラム ( PDB タンパク質 - 4 サブユニット )。
このプログラムは 各サブユニットに対し 約 500 アミノ酸まで扱える ( 500 × 4 = 2000 )。
DNA, RNA 部分は 独立したサブユニットとして扱われる。
( そのため 例えば 1 × DNA + 3 × タンパク質サブユニットなど )。
"HETATM" 部分 (= 通常のタンパク質以外 ) は その直前のタンパク質サブユニット内に組み込まれる。
2量体タンパク質 ( PDB 2J96 ) テキストファイル。
このタンパク質は 光感受性であり、2つの短いタンパク質サブユニットからなる
( 162 アミノ酸 × 2 )。
そのため このタンパク質からスタートするのが適切と思われる。
このテキストファイルを "C:\sdff" のフォルダーに ファイル名 "2J96.txt" でセーブする。
( もしくは このタンパク質のテキストを PDB ウェブサイトから直接セーブしてもいい。)
その後、上記のサンプル JAVA プログラムを "pf.java" としてセーブしてコンパイルしてほしい。
"詳細は、-Xlint: unchecked オプションを指定して再コンパイルしてください。" などの注意は無視してそのまま実行できる。
(Fig.2) プログラム内のセーブフォルダー位置の変更。

このテキストエディタを別のフォルダーにセーブしたときは、上記プログラム内のこのテキスト部分を変更するように。 ( Fig.2 の青線部分を変更させる。 )
タンパク質相互作用構造や 原子間距離のみを知りたいときは、
"coulom" の値をゼロにする。
( int coulom = 0 ).
この部分を " coulom= 1 " に変更すると、 クーロンポテンシャルや力が このページのように計算される。
しかし クーロン力の計算は スタートアップに 少し時間がかかるため、最初に
"coulom=0" の状態を選ぶ。
(Fig.3) コマンドプロンプト画面。

プログラム実行後、短い原子間距離が画面上に表示される。
O-C、 O-N、 O-O、 N-C、 C-C、 N-N などの原子間距離が 2.6 Å 未満のとき ( mixed subunit では 3.0 Å 以下 )、この原子位置をプログラム原子番号で示す。
例えば、 THR-N.39=THR-O.42>2.4536 は トレオニンの窒素 (= 原子番号, 39 ) と トレオニンの酸素 (= 42 ) 間の距離が 2.4536 Å であることを示している。
(Fig.4) 窒素における相互作用の固定部分。

このページで述べたとおり、Fig.4 に示すように
各原子近くの固定部分は除いて計算される。
例えば、Fig.4 の窒素のケースでは、N 原子と 線で囲まれた内部の原子間距離は C や CA における回転によって変化しない。
これはつまり このエリア外の原子が 窒素近くの各二面角を決定していることを意味している。
(Fig.5) N-3 と N-4 間の相互作用。

Fig.5 では、 窒素 3 と 4 ( N-3 と N-4 ) 間の距離が それらの間の二面角に影響を与えている。
一方、 N-3 と CA ( もしくは C ) 間の距離は この回転によって 変化しない。
そのため CA や C は 固定エリア内にあり、N-4 は 外部にあることになる。
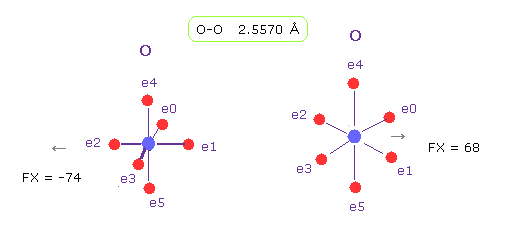
(Fig.6) O-O 長 = 2.6 Å
| O - O 長 | 5.0 Å | 4.5 Å | 4.0 Å | 3.5 Å | 3.0 Å | 2.5 Å | 2.0 Å |
|---|---|---|---|---|---|---|---|
| O1 of ( O - O ) | 19 | 23 | 30 | 39 | 53 | 78 | 125 |
| O2 of ( O - O ) | 19 | 23 | 29 | 38 | 51 | 71 | 97 |
| Average force | 19 | 23 | 29.5 | 38.5 | 52 | 75 | 111 |
このページに示したように、 このプログラムを用いて、2つの酸素原子核間の反発力 ( 力の単位: e+ と e- 間の距離がボーア半径のとき "1000" ) は その O-O 間距離が 2.6 Å 以下のとき 非常に強くなる。
そのため この距離 (= 約 2.5 Å ) は O-O ( もしくは O-N ) 非共有結合の 下限極限あたりと考えられる。
実際に Fig.3 では、 約 2.5 Å が下限である ( 2.4 Å 未満の距離は 各サブユニット内で 見られない。 )
同じことが サブユニット A と B 間の距離について言える。
( Fig.3 に示したように、2.4448 Å の O-O 長が A-B タンパク質の距離の 下限である。 )
(Fig.6) タンパク質画面。

基本的な操作方法は このページとほぼ同じである。
このプログラムには いくつか変更点がある。
前のバージョンのように、矢印 (= a ) と 円 (= b ) は "移動" と "回転" を行う。
x-y 平面上の ×+円 マーク (= c ) をクリックすると、すべての分子は -z 方向へ移動する。
( x-y 平面上の 点+円 マーク (=d ) をクリックすると、 z 方向へ移動する。 )
(Fig.7) テキスト画面。

この新しいプログラムでは、3種類の原子番号を使用している。
最初が プログラム原子番号で、 これは 水素原子を含むすべてに与えられる。
2番目が PDB 原子番号、 3番目が PDB 残基 (アミノ酸) 番号である。
( 水素原子は PDB 原子番号を持っていない。 )
(Fig.8) PDB データファイル。

"number" ボタンをクリックすると、画面上に表示されている 番号の種類を変更できる。
( 個人的には PDB 残基番号が見やすいので試してほしい。 )
文字 "B" が B サブユニットの原子にくっつく。
(Fig.9) "ang colo"、 "zoom"、 "x-y,x-z" ボタン。

プログラム実行後、画面上には 非常に多くの原子が同時に表示されるため どんな状態か分かりずらい。
そこで まず "60" の値を "zoom" ボタンの横に入力して それをクリックする。
それから "x-y,x-z" ボタンを2回クリックする。
サブユニット A と B タンパク質間の構造関係が見たいため、 "ang colo" ボタンをクリックする ( Fig.7 参照のこと )。
"ang colo" ボタンクリック後、 サブユニット A 原子は 赤色、 B は緑、 C は ピンク、 D は黄色になる。
( HETATM 原子は 紫になる。 )
すると 容易に サブユニット A と B のタンパク質を見分けることができる。
(Fig.10) サブユニット A と B。

次に A と B のタンパク質が どのように互いにくっついているか調べるために タンパク質全体の 断面図を表示させる。
(Fig.11) x-width を指定する。

Fig.11 のスクロールバーから "+-" を選んで、 "5" (= 5.0 Å ) の値を distance の横に入力し "x-width" ボタンを
クリックする。
この状態では、指定の部分の x 方向に 5.0 Å の幅を持つ 断面図が y-z 平面 (= 右画面 ) に表示される。
(Fig.12) x-width を指定する。

画面上のある粒子をクリックすると、この粒子に関する情報があるテキストボックスにジャンプする。
このページで述べたように、 "0" の値を "to=" ボタンの横に入力した状態で ある粒子をクリックすると、この粒子は "selected" (= 選択 )原子になる。
( "0" 以外の値を入力した状態で クリックすると、単にその粒子のテキストボックスへジャンプするだけである。)
"dis (A)" の行は 各原子と 選択原子間の距離を表示している。
見て分かるとおり、基本的に バリンやロイシンなどの疎水性のアミノ酸が A と B サブユニット境界付近に並んでいる。
そして 端付近に 各チロシンが 別のサブユニット内に食い込んでいる。
( これが これら2つのサブユニットが 互いにくっついている理由である。)
(Fig.13) マーク もしくは 原子の固定。

"mark fix" の状態では、ある矢印 ( もしくは x や 点マーク ) をクリックすると、その方向へ タンパク質全体が移動する。
( その移動距離は オングストローム単位で "distance=" 横の数値分である。)
"Fix" ボタンをクリックすると、 "all-free" や "atom-fix" の条件に移る。
"atom-fix" 条件では、タンパク質の代わりに マークのみが その方向へ移動する。
また "all-free" の条件では、 マークとタンパク質両方が移動する。
Fig.11 で スクロールバー内から "all" を選んで "x-width" ボタンをクリックすると、すべての原子が表示された元の状態に戻れる。
この新しいプログラムは DNA, RNA - タンパク質複合体も扱える。
基本的に DNA ( もしくは RNA ) のグループは 1サブユニットとして扱われる。
ここでは 次の PDB タンパク質を用いて説明する。
このテキストをセーブ ( もしくは PDB サイトから直接 ) して、JAVA プログラム内の 指定ファイル名を 1TGH に変更する。
(Fig.14) 原子と残基。

上のテキストファイルでは、 DNA 部分が最初に現れ、次に タンパク質 + HETATM 部分が続く。
このケースでは、 "protein+HETATM" 部分が サブユニット A として扱われ、 DNA 部分が サブユニット B として扱われる。
( なぜなら DNA 部分は短く、タンパク質部分が重要だからである。)
(Fig.15) 表示される残基を選択。

基本的に サブユニット B を選択するとき、A と B を区別するために 元の数値に
"10000" をつける。
例えば、サブユニット B の PDB 残基 112 と 113 のみを表示させたいときは、
値 "10112" と "10113" を "from"、 "to" のテキストボックスに入力して "display" ボタンをクリックする ( スクロールバー内から "residue" を選択後である )。
なぜなら A と B 両サブユニット内で 同じ残基番号が使用されていることもあるからである。
一方、PDB 原子番号は 各原子に固有のものであるため、"10000" などは PDB 原子番号につける必要はない。
(Fig.16) 112 (= G ) と 113 (= C ) の残基。

結果的に、112 と 113 の残基 ( もしくは 塩基 ) のみが画面上に表示される。
( PDBファイルでは 水素原子は不足していることが多いため このプログラムでは 自動的に新しい水素原子を付加している。)
(Fig.17) 原子番号 (= number )。
この新しいプログラムでは、3種類の原子番号を使用している。
最初が プログラム原子番号で、 これは 水素原子を含むすべてに与えられる。
B サブユニット (もしくは DNA ) に、 文字 "B" が 上のテキストボックス内に付けられる。
このプログラム原子番号を "select"、 "display"、 "from-to" のテキストボックスで指定するときは、 値 "10000" (= B サブユニット )、 "20000" (= C サブユニット )、 "30000" ( D サブユニット ) を 区別のため それに足す必要がある。
例えば C サブユニットの 数 4 は "20004" で指定する。
上のテキストボックス内の 2列目 (= PDB ) では、 PDB 原子番号と 残基番号が表示される。
PDB 原子番号 ( 例えば p2350 ) は 各原子に 特異的であるため、 この PDB 番号に "10000" などの数値を付加する必要はない。
そのため スクロールバーから "PDBat" を選択して "1" の番号を "select" ボタンの テキストボックス内に入力して それをクリックすると、 PDB 番号 "1" の原子 (= DNA シトシン の炭素 C5' ) が "選択原子" になる。
( この PDB番号を使って 水素原子を選択することはできない。)
2列目には PDB 残基 ( アミノ酸 ) 番号 も含まれている ( 例えば b507 )。
基本的に 異なったサブユニットにおいても 同じ残基番号が使われている。
そのため この残基番号を指定するときは、B-D サブユニットには それぞれ
10000-30000 の数値を 足す必要がある。
例えば、C サブユニットの PDB 残基番号 "5" のみを表示させたいときは、"from" と "to" のテキストボックス内に "20005" と "20005" を入力して "display" ボタンをクリックする。
"number" ボタンクリックすると、画面粒子上に表示される原子番号の種類を変更できる。
基本的に 残基番号が 短くて見やすい。
( また PDB 原子番号では 水素原子上に番号が表示されないため これも見やすい。)
(Fig.18) テキストボックス内の表示原子を変更する。

"Atom" ボタンをクリックすると、 次の原子の列 ( 5 - 9 ) が表示される。
"+Z back-1" ボタンをクリックすると、1ページ前の列に戻る。
"+Y back" ボタンをクリックすると、 最初の原子の列 (= 0 - 4 ) に戻る。
"+X (A)" ボタンをクリックすると、選択した原子に対する相対座標が表示される。
"dis (A)" は 選択された原子と 各原子間の 距離である。
( このケースでは 0-N と 1-CA の距離は 1.473 Å である。 )
(Fig.19) 選択原子。

スクロールバー内から条件 ("atom" もしくは "PDBat") を選んで、"select" ボタン横のテキストに数値を入力して そのボタンをクリックする。
Fig.19 のケースでは、 プログラム原子番号 "40" (= トレオニンの炭素 CA ) が "選択原子" となる。 ( "*" のマークが付く。 )
"dis (A)" の列では、各原子と選択原子間の距離が表示される。
同様に、条件を選択後に 原子番号を "to" の横に入力して "to" ボタンをクリックすると、指定原子のテキストボックスへ ジュンプする。
" | " マークが "to" 原子に付加される。
この "to" 原子は 選択原子と異なり、単に テキストボックスへ移動するだけのものである。
Fig.19 で、 "PDBat" をスクロールバー内から選択していれば、"40"、"41" の代わりに "19" もしくは "20" を 各テキストボックス内に入力する。
(Fig.20) 画面上の原子を直接クリックして "選択原子" を変更する。

画面上の ある原子を直接クリックして 選択原子を変更できる。
例えば、"46" の赤の円 (= 窒素) をクリックする。
この時点で、"to=" 横のテキストボックス内に "0" が入力されている状態だと 選択原子が "46" に変更される。
( "to=" ボタンはクリックする必要はない。ただ その横に "0" を入力するだけでいい。 )
もし "to=" 横に "0" 以外の数値を入力した状態で "46" 原子の丸をクリックすると、選択原子は変更されないまま、"46" 原子のテキスト画面にジャンプする。
( つまりこのケースでは "*" のマークが "46" 原子につかない。)
(Fig.21) x-width の指定。
Fig.21 で スクロールバー内から "+-" を選択して "5" (= 5.0 Å ) の数値を distance 横のテキストボックス内に入力して "x-width" ボタンをクリックする。
この状態では、Fig.22 に示したように x 方向へ 幅 5.0 Å の部分が y-z 平面上 (= 右画面 ) に表示される。
(Fig.22) x-width の指定。
スクロールバー内から "+" を選んでいれば 指定場所から x 方向に 0 から +5.0 Å の幅が表示される。
( "-" の場合は、 -5.0 Å から 0 の範囲となる。 )
注意: 例えば x-z 平面は 必ず x、z 方向の すべての原子を表示する。
そのため Fig.22 で "z width" を指定しても 画面は変化しない。
( なぜなら x-z と y-z の両方とも z 方向を含んでいるからである。)
上記のスクロールバー内で "all" を選んで、 x-width ( もしくは y, z- width ) ボタンをクリックすると、元の状態に戻れる。
(Fig.23) Mark もしくは atom fix.
"mark fix" の状態では、ある矢印 ( もしくは x や 点マーク ) をクリックすると、その方向へ タンパク質全体が移動する。
( その移動距離は オングストローム単位で "distance=" 横の数値分である。)
Fig.23 では、矢印の1回クリックにより タンパク質全体が x 方向へ 2.0 Å 移動する。
"Fix" ボタンをクリックすると、 "all-free" や "atom-fix" の条件に移る。
"atom-fix" 条件では、タンパク質の代わりに マークのみが その方向へ移動する。
また "all-free" の条件では、 マークとタンパク質両方が移動する。
(Fig.24) "ang colo"、 "zoom"、 "x-y,x-z" ボタン。
"zoom" ボタン横のテキストに数値を入力して そのボタンをクリックすると、タンパク質のズームが変化する。
"100" の値が最初の状態。
"200" を入力すると、 タンパク質全体が 2倍に拡大する。
"50" を入力すると、タンパク質全体が 元の像の半分に縮小する。
( 最初に タンパク質全体像を把握するために 縮小させるのがいい。)
"x-y、x-z" ボタンをクリックすると、左右の座標画面を変更できる。
最初が "x-y, x-z" 平面、 2番目が "x-y, y-z" 平面、 3番目が "x-z,y-z" 平面である。
"ang colo" ボタンをクリックすると ( Fig.17、 Fig.21 参照 )、サブユニットA が 赤色、 B が 緑色、 C がピンク、 D が黄色になる。
( また HETATM 原子は 紫になる。 )
そのため A と B のタンパク質サブユニットを容易に区別し その相互作用構造を把握できる。
(Fig.25) "center" ボタン。

ある値 ( 例えば "5" ) を "distance" 横の テキストボックス内に入力して"center" ボタンをクリックすると、選択原子から 5.0 Å 以内の距離の原子のみ画面に表示される。
( この新しいプログラムでは、"nucleus" 原子という概念はない。)
初期値は 80 Å である。
この "center" と "width" ボタンを組み合わせれば、タンパク質の様々な部分の構造が 容易に理解できる。
このボタンをクリックすると、他のサブユニット近くのエリアのみが表示される。
"3.5" を入力すると、A と B ( もしくは B と C ) の間の境界 "3.5" Å 付近に含まれる原子のみ表示される。
このボタンは すべての距離を計算するのに 少し時間がかかる ( そのため あまりこのボタンは使用しなくていいと思われる。)
この 3.5 の値を 他の値に変えたとき、"AB-border" から "all" への変更過程で この条件に移る。
そのため この変更効果を確認するには このボタンを 2,3 回クリックする必要がある。
(Fig.26) "subunit" ボタン。

スクロールバー内から "erase" を選択して、 "1" をテキストボックス内に入力して "subunit" ボタンをクリックする。
この場合では サブユニット B のタンパク質が 画面から消える。
"0" を入力したとき、サブユニット A が消える。
( 0-サブユニット A, 1-サブユニット B, 2-サブユニット C, 3-サブユニット D. )
スクロールバー内から "all" を選択して、 "subunit" ボタンをクリックすると、元の状態に戻れる。
(Fig.27) 2つの任意の原子間の距離。

coulom=0 の条件で、2つの粒子を続けてクリックすると、それらの原子間距離がテキストボックス内に表示される。
Fig.27 では、最初に メチオニンの N 原子 (= 原子番号 0 ) をクリックし、次に H2O の酸素原子 (= B サブユニット 2722 ) をクリックしている。
これらの数値はプログラム原子番号である。
Fig.27 のケースでは、O の原子が選択されており、それが 四角形になっている。
また直前にクリックした原子は 黄色の四角形になる。
( もちろん、"dis A" の列を見ても、各原子と選択原子間の距離を知ることができる。)
他のコマンドは このページと ほぼ同じである。
少し変更箇所がある。
"from-to-display" コマンドでは スクロールバー内から "residue" の条件のみ選択できる。
例えば、"from" と "to" の横のテキストに "1" と "5" を入力して "display" ボタンをクリックすると、
サブユニット A の 残基番号 1-5 のアミノ酸のみ画面に表示される。
"10001" と "10005" をテキストボックス内 ( "from" と "to" の横 ) に入力して "display" ボタンをクリックすると、サブユニットB の残基番号 1-5 のアミノ酸のみが 画面上に表示される。
"C" と "D" のサブユニットに関しては、"20000" もしくは "30000" を 残基番号に付加する。
"dihedral"、 "chain rot" ボタンに関しては、 このページ とほぼ同じである。
サブユニット B 内の 二面角を知りたいときは、 "10000" を プログラム原子番号に付加する。
( PDB 原子番号に "10000" をつける必要はない。 )
基本的に 1つのタンパク質サブユニットは "TER" で終わる。
"TER" に続くアミノ酸は 次のサブユニットとなる。
"HETATM" の部分は 直前のサブユニットに吸収されてカウントされる。

2013/8/8 updated This site is link free.