
(Fig.1) Success rates of designing de novo proteins bound to known target peptides are too bad (= 6 % ~ 50 % ) to use for drug development. ↓ by Nature
Due to the current useless mainstream protein simulating methods such as quantum mechanical density functional theory (= DFT ) and time-consuming molecular dynamics (= MD ), researchers can Not know the precise atomic interactions nor design effective drugs, proteins.
Even the latest tools for designing and predicting proteins bound to some known target peptides have so bad a success rate that they cannot be used for developing effective drugs or predicting precise side effects.
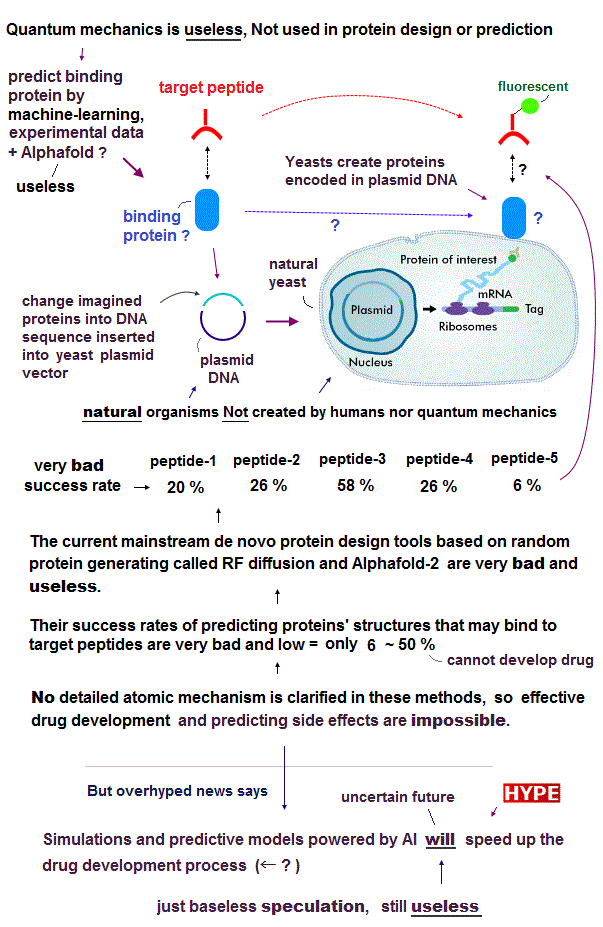
According to the latest (exaggerated AI) research published in Nature, the current mainstream de novo protein design method just tried to add randomly-generated noises or random amino acids to new proteins against some known target peptides (which method is called RF diffusion, this 7th-paragraph, this p.5-Peptide binder design ) combined with the Alphafold-2 (= AF2 ) predicting protein structure, and other alleged machine-learning methods (= ProteinMPNN, Hallucination, RFjoint inpainting ). ← No (useless) quantum mechanics is used.
They inserted DNAs allegedly encoding these new proteins designed by the random RF-diffusion and Alphafold-2 into yeast plasmid DNA vector, and get (natural) yeasts to generate those artificially-designed proteins on their cells' surface.
If their protein design is right, those newly-produced proteins could bind to target peptides labeled with fluorescent (= biotinylated ) substances in screening using yeast displaying proteins ( this p.1, this p.5-left-2nd-paragraph ). ← Precise atomic interactions between proteins or where exactly of these new proteins bind to peptides are unknown, though.
In these methods, they designed and produced 96 new different binder proteins for each (natural known) target peptide such as neuropeptide Y (= NPY ), glucagon (= GCG ), parathyroid hormone (= PTH ), and apoptosis peptide called Bim ( this 4th-paragraph ).
Success rates of new designed proteins bound to target peptides are very bad = only 20% (= 20/96 ) in NPY, 26% (= 25/96 ) in GCG, 58% (= 56/96 ) in PTH, 26% (= 25/96 ) in Bim.
See this p.7(or p.6)-2nd-paragraph,p.8(or p.7)-2nd-paragraph, or this p.6-1st,3rd-paragraphs,
In the case of designing protein binders to secretin (= SCT ) peptide, its success rate is worst, only 6 % (= only 4 proteins bound to the target SCT peptides out of total 66 proteins designed to bind to SCT peptide, this p.5(or p.4)-1st-paragraph, or this p.4-1st-paragraph )
↑ Only 20% successful protein prediction rate, which cannot be used for effective drug development or predicting exact side effects.
↑ To precisely predict side effects, interactions with many other peptides, proteins, substances inside bodies must be precisely predicted, which is impossible in this very bad-success-rate RF-diffusion + Alphafold-2.
Furthermore, all these binding screening biological tools are based on natural organisms such as yeast's protein production system, and (viral) plasmid vector, which are Not designed nor created by humans from scratch due to useless quantum mechanical atomic theory.
These biological tools just seeing if new proteins may vaguely bind to target peptides just by seeing blots or fluorescence can Not clarify detailed atomic mechanisms, and even whether new proteins really bind to target peptides is unknown in this screening method.
↑ Due to the useless quantum mechanical atomic theory, researchers can only randomly create new proteins out of almost infinite candidates and see if they may bind to target peptides by very time-consuming trial-and-error approaches without atomic-level prediction, which almost-blind trial-and-error approach takes unrealitically too much time to develop drugs.
Microsoft's ad-hoc protein diffusion model (= RF diffusion, Evodiff, Diffdock.. ) also just tries to design and generate somewhat (meaningless) random (noise) proteins' sequences and structures against some target molecule (= static useless protein like Alphafold ) by (overhyped dubious) AI (= Not quantum mechanical prediction, this 7th-paragraph ), which successful protein binding prediction rate is still impractically low (= only less than 19% even with the help of Alphafold-2, this p.8-Design of protein biding, p.9, Fig.6b, this 9th-last paragraph, this p.3-last-paragraph ), and lacking many experimental verification ( this 8,13th paragraphs, this-lower Conclusion, this 7,11th-paragraphs, this p.1,p.2-right ), so it's also useless for drug development contrary to the media-hype.
↑ These are typical examples preventing real scientific development or curing diseases by overhyped AI that just wastes huge taxpayers' money and time only in these fruitless science (= whose only purpose is to publish papers in top journals instead of curing diseases ) which just makes people wrongly believe the present science may be progressing (= actually Not ).
The 1st, 4th, 5th, last paragraphs of this hyped news say
"group at the University of Washington, have developed a novel deep learning method, RoseTTAFold All-Atom (RFAA), for prediction and design of complexes of proteins, small molecules, and nucleic acids. Subsequently, they developed RFdiffusionAA, which builds protein structures around small molecules (= still No ability to predict proteins accurately, though )."
"It was already known that bilins are optically featureless unless they are held within a defined binding site, at which point they become intensely colored and emissive. In this work current Ph.D. student.. devised a multiwell assay that could screen many RFdiffusionAA-generated genes in parallel, using E. coli cells that could make phycoerythrobilin (PEB)."
"Morey-Burrows evaluated 94 designs in one go, with the multiwell assay revealing visibly colored cells in nine wells (= only 9 proteins out of 94 proteins designed by deep learning seemed to bind to the target bilin, very bad prediction rate and useless for drug development ). He had identified nine proteins dissimilar to each other and to any native bilin binder, based on pigmentation or fluorescence."
"fluorescent reporter probes with tunable excitation/emission maxima would (= uncertain future, still useless ) be useful biochemical tools."
↑ This research tried to predict proteins binding to target molecules (= such as bilins ) by using the deep learning ( or AI ?) tool called RoseTTAFold All-Atom (RFAA) trained on experimentally-obtained structure data such as PDB (= useless quantum mechanics was Not used ), and its successful prediction rate was only 9.6% (= only 9 proteins out of 94 designed proteins barely bound to the target bilins ), which low success rate cannot be used for actual drug design that needs precise prediction of many proteins' interactions to avoid serious side effects.
↑ This research paper ( this ↓ )
p.1-Abstract says "AlphaFold2 (AF2) and RoseTTAFold (RF).. they are unable to model covalent modifications or interactions with small molecules and other non-protein molecules that can play key roles in biological function"
p.4-Training RFAA says "From the PDB, we curated a protein-biomolecule dataset including protein-small molecule.. we supplemented the training set with small molecule crystal structures from the Cambridge Structural Database" ← They trained or did "deep learning" based on experimentally-obtained protein, molecule database, Not on (useless) quantum mechanics.
p.10-2nd paragraph says "Starting from a panel of 94 RFdiffusionAA designs and using phycoerythrobilin (PEB) as the chromophore, we identified nine bilin-binders based on visible pigmentation or fluorescence in a whole cell screen (a 9.6% hit rate )" ← This deep-learning successful rates of predicting proteins binding to the target bilin molecules is very bad, only 9.6% = 9/94.
↑ To deveop or find effective drugs, we have to precisely predict interactions between drug molecules and many proteins inside bodies to avoid the serious side effect, so this extremely-low prediction rate of protein-molecule interaction (= only 9.6% ) can Not be used for practical drug design.
The point is these deep-learning or AI methods do Not consider the detailed atomic or molecular interactions, instead, they just tried to train AI tools based only on the experimentally-observed (final static) protein or molecule structures (= without considering the "process" of how each protein was folded through atomic interaction due to the current mainstream physics unable to give realistic "shapes" to atoms ).
This is why the current biological and medical researchers are still unable to utilize practical atomic interactions for drug development, and they just waste their time only to publish papers in academic journals whose useless pseudo-science ruins their careers and future.
The 3rd, 6th, 11th, last paragraphs of this hyped news says
"The authors say technique is accurate, fast, cost-effective and has the potential (= just speculation, still useless ) to revolutionize drug discovery "
"the accuracy of AlphaFold 2 has revolutionized protein structure prediction, the method has limitations: It allows scientists to model proteins only in a static state at a specific point in time (= Alphafold2 cannot predict protein's conformational change, this 2nd-paragraph )."
"In this study, the researchers were able to manipulate the evolutionary signals from the protein to use AlphaFold 2 to rapidly predict multiple protein conformations, as well as how often those structures are populated (= based on experimentally-obtained data, and Alphafold2 or AI cannot deal with protein interaction nor drug discovery, this 5-6th-paragraphs )."
"As for next steps (= uncertain future, still unrealized ), the research team is refining their machine learning approach.."
↑ This research tried to roughly estimate probabilities (= populations ) of generating some protein conformations using Alphafold2 based on experimentally-obtained data (= No quantum mechanical prediction ), and failed in predicting various protein conformations.
This research did Not consider protein or drug interaction (= this research focused only on proteins' vague probabilites or populations ), so this research or Alphafold 2 remains useless for predicting protein interaction or drug discovery.
Furthermore, this research just (almost) randomly sampled multiple protein conformations, and did Not explain how proteins change their conformations nor its mechanism.
The basic protein or atomic mechanism remains unknown and unresolved due to useless quantum mechanics.
↑ This research paper ↓
p.1-right-1st-paragraph says "AlphaFold 2 was trained using large amounts of experimental data (= Not quantum mechanical prediction )"
p.1-right-last~p.2-left says "AF2's (= Alphafold2's ) inability to predict multiple conformations is unsurprising"
p.2-right Results-1st-paragraph say " AF2 takes as input a target sequence and a corresponding multiple sequence alignment (= MSA ). An arbitrary number of sequences (defined by the max_seq parameter = artificially-optimized parameters, this p.4-1st-paragraph ) are randomly selected from the master MSA (= true protein mechanism is unclear )"
p.2-right Results-2nd-paragraph says "We chose the Abl1 kinase core. Abl1 is thought to occupy three major conformations with different populations (← this research's Alphafold2 tried to predict only thse populations, No protein interaction considered ). Abl1 primarily exists in an active (ground) state. Infrequently, Abl1 will switch to inactive state 1 (I1), and then to inactive state 2 (I2)"
p.4-left-1st-paragraph says "none of the Src (= kinase ) predictions were found to be in the I2 state, although
the enzyme is known to infrequently occupy this conformation. This
suggests a resolution limitation in using AF2 to predict relative state
populations (= Alphafold2 failed to predict )"
p.8-left-2nd-paragraph says "our method failed
to replicate the significant changes in dynamics of residues 80–90 for
the H83Y mutations (= prediction failed )"
p.10-left-3rd-paragraph says "our workflow inaccurately predicted the M290L mutation "
p.10-left-last-paragraph says "the A380 side-chain ranges from −130
(active state) to 40 (inactive state) degrees in structures in the Protein
Data Bank (PDB)23, but ranges from −130 to −90 degrees in AF2 predictions (= Alphafold2 failed to predict protein structure, too )."
As a result, Alphafold2 or AI remains unable to explain protein intearaction or conformational change (= its prediction of protein structures still often failed ), hence, drug discovery is impossible in contradiction to the repeated hypes ( this 3rd-paragraph ).
It is impossible for computer's simulation such as AI and machine learning to contribute to useful drug development as long as they rely on the current ancient impractical quantum mechanics lacking real atomic picture with shapes, as shown in the upper section.
In order to hush up this fact of hopeless AI, many corporations, academa, journals and the media need to spread baseless overhyped news.
For example, the middle of this hyped news says
" Some people predict that drug development will (= just uncertain future, still useless ) be transformed by AI..(?)"
"Simulations and predictive models powered by AI will (= just baseless speculation ) speed up the drug development process (?)"
The 5th-paragraph of another hyped news says
"Near term, we believe AI will (= uncertain future, still useless now ) usher in a new era of drug discovery (?)"
The 1st paragraph of this hyped article says
"Machine learning and other technologies are expected to (= just baseless speculation ) make the hunt for new pharmaceuticals quicker, cheaper and more effective (?)"
The last paragraph of this hyped news says
"No longer will (= just speculative future, nothing is realized yet ) it take just ML and AI to play in the drug discovery and development sandbox—it’s going (= uncertain future, again ) to include molecular simulation too. "
↑ None of these hyped news talking only about "uncertain speculative future" states that AI actually produced anything useful, nor specifies what they actually did in detail.
So the real fact is the current AI with imaginary promise achieved nothing practical.
Artificial intelligence (= AI ) and machine-learning are overhyped useless pseudo-technology created by various global companies and academia to cover up the inconvenient fact that the present science and innovation miserably stop progressing stuck in fictional, impractical mainstream science, as shown in still-impractical self-driving cars.
Even the recent hyped AI research allegedly mimicking human brain intelligence using ordinary (synaptic) transistors (= useless quantum computers are Not used ) did Not reach human brain at all.
All they did in this hyped research published in Nature was just get their computer transistors to recognize only three bitstring = 000. ← No more complicated tasks were performed ( this-4th-last-paragraph, this 11th-paragraph ).
So even this latest alleged-AI technology is far from real human intelligence (= due to useless quantum mechanical atomic theory preventing researchers from using real atomic interactions ).
This exaggerated AI research also did Not unravel mysteries or precise mechanisms (= of some dislocation ) of materials at all, becaue of their reliance on the unrealistic one-pseudo-electron DFT model ( this p.12-left-2nd-paragraph ).

2023/12/20 updated. Feel free to link to this site.
{kind=link}
{kind=link}
{kind=link}