
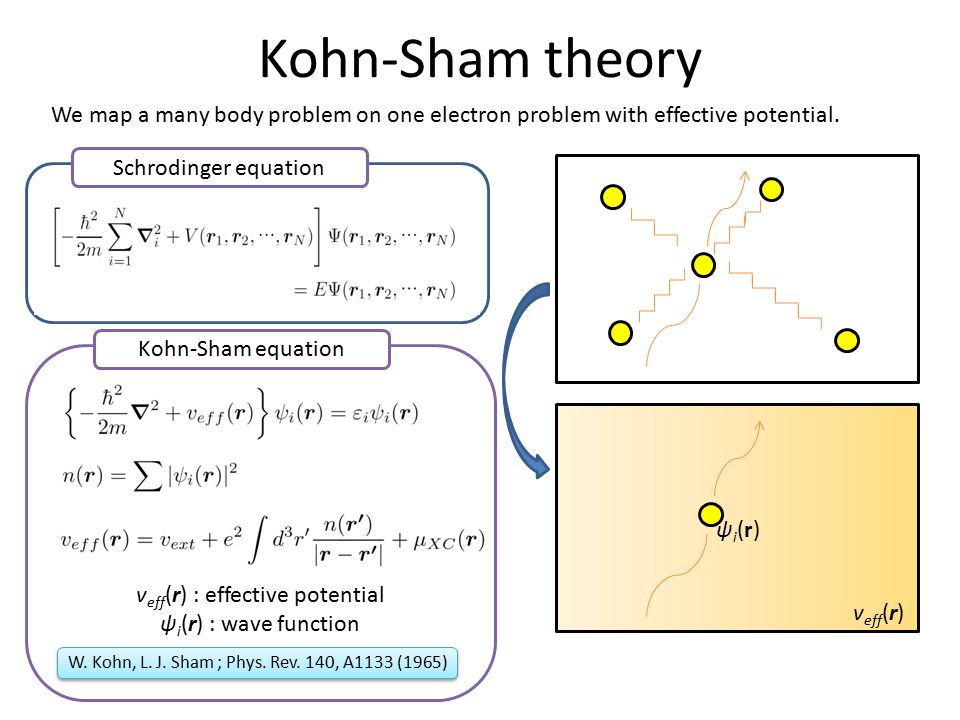
(Fig.1) Today's only mainstream quantum mechanical approximation = density functional theory (= DFT ) is unreal, hampering science
Quantum mechanical Schrödinger equations are useless and unsolvable (= so cannot predict energies ) for any multi-electron molecules and materials.
Physicists have to artificially choose fake trial wavefunctions or basis sets with a lot of freely-adjustable parameters, which is impractical, taking too much time to deal with multi-electron Schrödinger equations ( this p.2-3, this p.11, this 2.22 ).
Quantum mechanical unphysical Pauli principle requires these artificially-chosen (fake) trial wavefunctions to take antisymmetric forms (= composed of terms exchanging electrons ) where every single electron needs to be in all different atoms and orbitals simultaneously, which unrealistically spreading single electron forbids each atom from having its clear boundary or shape.
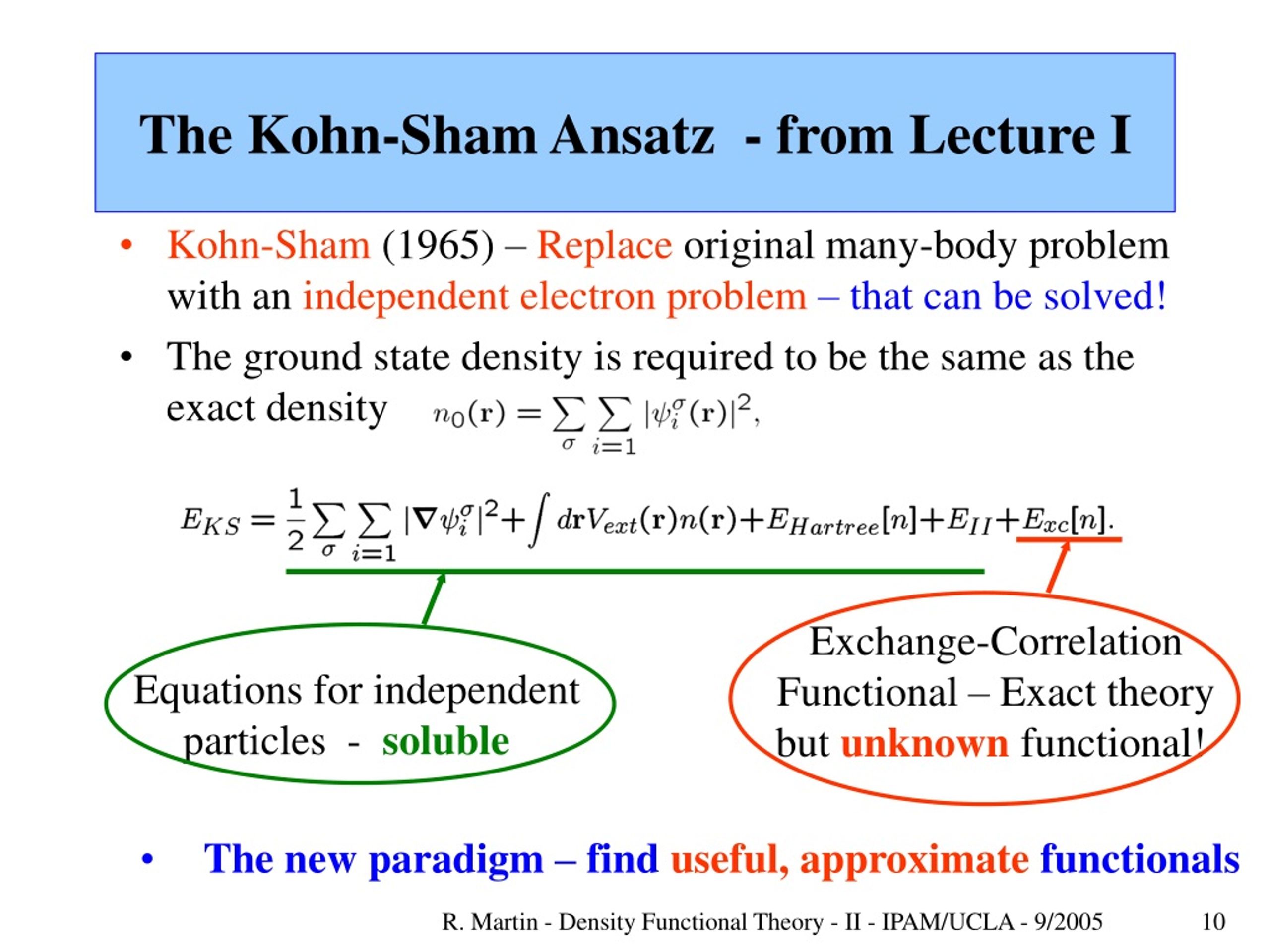
This unphysical inconvenient quantum mechanics forced physicists to rely on (unrealistic) approximation called density functional theory (= DFT = Kohn-Sham theory ) that unreasonably treats any multi-electron molecules or materials as one pseudo-electron (= density ) or fictional quasiparticle model ( this p.3-5, this p.2-1.1 ).
In order to express multi-electron molecules and materials, DFT uses only one fictitious electron ( this p.27 ) or only one particle's coordinate ( this p.3-4 ) that can Not give boundaries or shapes to individual atoms.
Of course, this DFT's one-pseudo-electron model is unrealistic, fictitious and has No physical meaning ( this p.17(or p.5)-2nd-last-paragraph, this p.2-1st~2nd-paragraph, this p.3-2.2, this-last-paragraph, this p.15, this p.21-3rd-paragraph ).
↑ This unphysical quantum mechanics or its DFT lacking real atomic shapes or physical meaning cannot be used as real atomic models.
DFT's one pseudo-electron causes the unrealistic self-interaction energy error (= due to one fictitious electron repelling itself by its own electric repulsion, this p.13-14 ), and always gives wrong (absolute) total energies.
This unphysical DFT is the current only mainstream quantum mechanical method used in all fields such as condensed matter, semiconductor, fictional quasiparticles, superconductor, biology. ← In other words, this unphysical quantum mechanical DFT lacking real atomic picture hampers all scientific fields.
In this DFT one-pseudo-electron method, physicists have to artificially choose fake energy called exchange correlation functionals ( this p.12-13 ).
The universally-correct exchange correlation energy functional is unknown ( this-introduction-1st-paragraph, this p.8 ), and different exchange energy functionals have to be created and chosen in different molecules and situations, each time they give wrong results ( this p.7, this p.17 ).
This p.1-intorduction-1st-paragraph says
"Te precision and reliability of DFT
computations are greatly influenced by the choice of exchange–correlation functional"
Furthermore, like unsolvable Schrödinger equations, DFT also has to choose or guess fake wavefunctions (= densities ) or basis sets with many freely-adjustable parameters in different molecules ( this p.11, this p.10 ).
↑ Dependence on artificially-chosen (fake) exchange energies, wavefunctions ( this p.4,p.9-right-3rd-paragraph ) and experimentally-obtained bond parameters ( this 3rd-paragraph ) means DFT is also unable to predict any atomic energies, hence, DFT is just an empirical, Non-ab-initio theory ( this p.23-lower, this p.11-left-1st-paragraph, this 7~8th-paragraphs, this p.2-last ).
This p.19(or p.31)-last-paragraph says
"Due to the approximate nature of the exchange correlation functionals in use today,
the absolute energies obtained from DFT are not inherently meaningful."
This DFT with unphysical shapeless atomic model has to conduct too time-consuming meaningless calculations, so quantum mechanics and DFT is an unnecessary equation just hampering nano-technology.
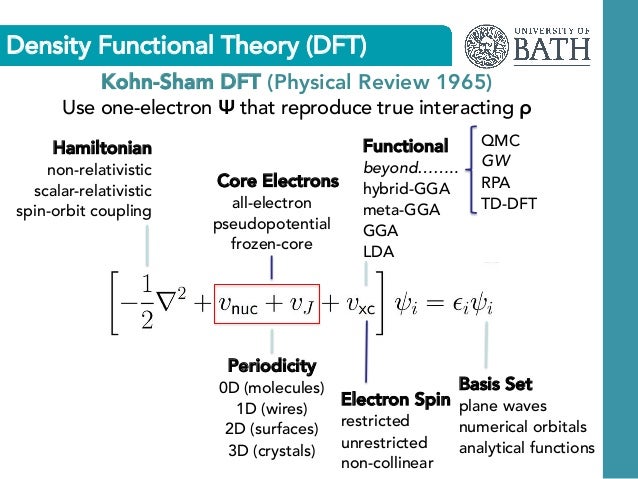
(Fig.2) Quantum mechanics and DFT must rely on rough approximation of pseudo-potential and Hubbard energy parameter U, which cannot predict atomic energy.

Due to useless Schrödinger equations, the current mainstream quantum mechanical one-fake-electron approximation = DFT (= Kohn-Sham theory ) also has to choose fictional pseudo-potential expressing atomic core electrons in addition to choosing the artificial exchange correlation energy functionals.
They can choose various different pseudo-potentials that often fail ( this p.14, this p.7-last, this p.2 ), and even full-potential method (= not pseudo-potential ) also has to artificially choose energy parameters, which cannot predict any physical values.
This or this p.3-last-paragraph (= how to create new pseudo-potentials ) says
"The second and third steps are closer to cooking than to science. There is a large
arbitrariness in the preceding step that one would like to exploit in order to get the
"best" PP (= pseudo-potential ), but there is No well-defined way to do this"
All DFT calculation packages such as VASP, Espresso, and projector augmented wave (= PAW ) depend on this artificial choice of pseudo-potential and various parameters ( this p.18 ), so they cannot predict any physical values.
The paradoxical relativistic effect such as spin-orbit interaction must be expressed by these artificial pseudo-potentials for relativistic version ( this 2nd~4th-paragraphs, this-p.2-4th~5th-paragraphs ), Not by quantum mechanical prediction.
DFT often uses unphysical plane wave (= basis set ) spreading over the whole material expressing one-pseudo-electron lacking real atomic shape ( this p.11-12, this-lower-Fig.5, this p.1-left-1st-paragraph, this p.8-DFT, this p.6-DFT ).
In metals, DFT is known to give wrong results, so it has to use and adjust additional free parameters called (empirical) Hubbard U or V parameters ( this abstract ).
This DFT-U or V additional potential parameters have to be artificially fitted to experiments ( this p.1-left-lower, this p.3-right ), which cannot predict any atomic energies.
(Fake) Ab-initio methods for determining this U parameters often depend on various artificial-chosen parameters such as pseudo-potentials ( this p.9-left-2nd-paragraph, this-lower-caveat ), which often give wrong results ( this p.3-3rd-paragraph, this p.2-left-1st-paragraph ).
This p.6-last says -- ab-initio U fail
"We also employed the ( ab-initio ) linear response method.. to determine a range
of U values; however, the values obtained by this method were too high ( this p.8-table I-headline )"
This-lower-6 summary and outlook-2nd-paragraph says
"the computed (= ab-initio ) U is Not necessarily being better than the empirical (= experimentally-determined ) ones." ← ab-initio U is incorrect
This p.2-3rd-paragraph (or this p.15-4. ) says -- Free U parameter
"(i.e., the Hubbard U and V). Unfortunately, the values of these parameters are not known a priori and it is still
quite a common practice in literature to evaluate them semi- empirically by fitting various experimental properties (when
available), which prevents this method from being fully ab initio and from being predictive for novel materials"
This-p.3-left-1st-paragraph says -- U cannot predict energy
"However, with such a correction, semi-empiricism in
the form of the two Hubbard parameters (or one effective
parameter Ueff = U−J) is introduced, which require a
system-dependent choice. Although rigorous ways of determining
the required parameters have been proposed,
the resulting parameters enable to reproduce often not all
the desired target quantities. For example, electronic
band gaps often turn out to be still severely underestimated
with such rigorously obtained Hubbard parameters. Thus, in
many studies this approach is not followed. Instead, Hubbard
parameters are adjusted by comparing results to a variety of
reference material properties (such as band gap, lattice or
spin structure, etc). Doing so, one usually finds that different
parameters are needed for reproducing different observables.
Such an approach has very limited predictive power"
Today's mainstream density functional theory (= DFT ) cannot explain band gap energy in various materials such as semiconductors ( this-p.37-39 ), which means quantum mechanics was useless for transistors.
This-p.1-left says -- DFT fails in band gap
"Despite its relative
ease of measurement, accurately predicting the band gap from first principles remains a challenging task, especially because interpreting the Kohn Sham (KS) gap from density functional theory (DFT)"
↑ DFT tries to artificially adjust pseudo-potentials and free energy parameters U to get band gap energy close to experiments, which ad-hoc methods cannot predict any actual energy (= DFT also cannot predict materials' absolute energy called Fermi energy ).
One ad-hoc quantum mechanical approximation tries to use fictitious quasiparticle-GW approximate method to artificially modify the wrong bad-gap energy obtained by the ordinary DFT with LDA exchange functional by the unreal quasiaprticle energy ( this-p.16 ).
↑ The problem is this GW method is Not legitimate nor self- consistent (= Non-SCF where values of initial guess and final results are inconsistent ), which means their calculated energy results heavily depend on the initially-chosen one-fake-electron DFT wavefunction, so the quasi-particle-GW cannot predict actual energy ( this-p.17(or p.11)-lower~p.18, ).
Because the self-consistent quasiparticle GW methods (= QSGW ) are known to give worse energy values than the illegitimate non-self-consistent one-shot G0W0 method ( this-p.17-left, this-p.1-right-1st-paragraph, this-p.2,p.29-right-2nd-paragraph, this-p.2-abstract-lower ).
This-p.3-right-1st-paragraph says -- Wrong quasiparticle-GW
"In the G0W0 approximation, the random phase
approximation is used, and the eigenvectors and eigenvalues
are used directly to construct G0 and W0. “0” thus refers to
“single-shot” calculations, which are non-self consistent (= Non-SCF )."
"this good agreement is largely due to the fortuitous cancellation of errors due to non-self-consistency and the lack of vertex corrections. Moreover, single-shot G0W0 may be sensitive to the starting positions (= non-self-consistent GW's results can be artificially adjusted by the initially-chosen atomic wavefunctions )"
"Although this can be overcome by full selfconsistency (scGW calculations), scGW often worsens the band gap by 10%"
The recent created quasiparticle-GW-EDMFT method tries to artificially separate orbitals into self-consistent and non-self-consistent (= one-shot GW ) ones to adjust band gap energies with the help of additional ad-hoc correction, which also cannot predict band gap energy due to various freely-chosen parameters ( this-p.1-right, this-p.17-left ).
To express intermolecular (= van der Waals ) energies, DFT has to choose empirical (= irrelevant to quantum mechanical prediction ) dispersion energy functionals called DFT-D, D2, D3 whose parameters must be fit to experimental results with No quantum mechanical prediction ( this p.4 ).
All other various forms of DFT's intermolecular van der Waals energy functional such as vdW-DF2, PBE-vdW.. often failed ( this p.6-right-3rd-paragraph, this p.3-last-paragraph ).
As a result, today's mainstream quantum mechanics and DFT are useless, unrealistic (= giving only unphysical one-pseudo-electron model ), unable to predict any molecular energies.
This quantum mechanical DFT one-pseudo-electron model lacking real individual atomic shape is a main culprit of hampering today's scientific development.
(Fig.3) Full-potential (= non-pseudo-potential ) DFT method (= FP-LAPW) just choosing artificial energy parameters E is also useless, unable to predict any physical values ↓

Not only artificially-created pseudo-potentials but also all-electron full potential methods are fake ab-initio, unable to predict any physical values such as atomic or band energies, and they have to artificially choose many free parameters such as energy parameters.
Because also in all-electron full potential method (= instead of pseudo-potential ). multi-electron Schrödinger equations are unsolvable (= cannot predict energy ) and impossible to apply, so physicists have to rely on unphysical one-pseudo-electron density functional theory (= DFT ) approximation with fake exchange energy functional and artificially-chosen potential energy, which is unable to give true potential energy or predict any values.
All-electron full-potential (= non-pseudo-potential DFT ) methods containing APW, LAPW, FP-LAPW (= full-potential-linearized augmented plane wave ) divide molecules or solids into two regions of atomic (sphere) core region and interstitial region containing valence electrons with unphysical plane wave (= No physical meaning, this p.3-2nd-last-paragraph ) and fictitious constant potential V (= fitting parameter, this p.3-left ).
So this all-electron full-potential method also has to artificially choose different radii of (fictional) atomic core (= called Muffin-tin or MT radius ) in different atoms, which is one of freely-fitting parameters ( this p.23, p.27-32 ).
Besides this atomic core radius, the all-electron full-potential (= non-pseudo-potential ) method has to artificially choose energy parameters (= El ) in different orbitals (= core, semi-core, local-orbitals, valence electrons' orbitals ), instead of finding these energy parameters by ordinary variational methods ( this-p.10-11,p.23-24, this p.6-12, this p.6-left, this p.44, this p.8,p.28-32, this p.29(or p.9)-(2.4), this p.20-lower ).
This-p.31(or p.29)-4.5.2 says -- Choose free energy parameter
"In order to minimize the linearization error, the energy parameters should be chosen as
close to the band energies as possible. However, the band energies ε(k) depend on k
whereas the energy parameters El
are constants. In addition, the radial functions contribute to the eigenfunctions of different band with different energies. Therefore, deviations between ε(k) and El have to be accepted.
This p.1-right-last~p.2-left says -- Full-potential needs free parameters
"However, approaching the complete basis limit using the FLAPW (= full-potential ) method still requires expert knowledge, e.g., of the atomic electronic structure in order to define the local orbital basis at appropriate energies, choosing parameters such as the MT radius and angular momentum cut offs, and controlling the linearization error."
This or this p.4-right-2nd-last-paragraph says -- choose energy parameters
"A clever choice of energy parameters Etℓ is
essential for accurate results, and WIEN2k has several automatic
ways to make an optimal choice in most cases
The energy parameters
of all other valence states are set to 0.2 Ry below EF. "
This p.92(or p.89)-2nd-paragraph says -- Adjust free energy parameter
"In the calculations the energy parameter is varied between −0.29 and 0.08 Htr (≈
between −7.9 and 2.2 eV) relative to the Fermi energy. Within this interval we observe that the conventional LAPW basis (= all electron, full-potential ) yields a strong dependence of the total energy on the choice of the energy parameter"
This p.4-2nd-paragaph says -- choose energy, radius
" Unlike the APW case, the KS secular
equation in the LAPW basis (= all electron ) is of the ordinary linear variational form. The only user-dependent choices are the reference energies and muffin-tin radii"
This p.4-left-2nd-paragaph says -- Fix free energy parameters
"The basis functions, defined in Eq. (16), can represent only those wave functions accurately whose energies
are sufficiently close to the energy parameters..
For semicore states, which are nearly dispersionless, the energy parameter is fixed at the semicore
energy level."
As a result, the present mainstream one-pseudo-electron DFT approximate method has to artificially choose pseudo-potential energy or choose energy parameters (= full-potential method, this-(5) ), which is useless, unable to predict any physical values.
This website says on full-potential DFT ↓
p.39(or p.36)-last-paragraph says -- Free energy parameter
"Assuming the spherical approximation, one can construct energy dependent basis functions ul(r,E ) within the muffin-tin spheres (= core region ) by solving the corresponding radial Schrodinger equation for an energy parameter Ek.
p.51(p.48)-3.1.3 Determination of the energy parameters-(3.15) mentions the occupation or the state of weight ωk which can artificially determine the probabilities of valance-electron band's (plane) wavefunctions with momentum (= or kinetic energy ) k influencing density ρ and total energy parameter E in p.52-(3.17).
p.52(or p.49)-4th-paragraph mentions -- Free energy parameter
"atomic energy parameters (= AFP)" which can artificially determine eigenenergies (= energy parameters ) of the states of an artificial (= fake ) atomic problem, which consists of the
spherical part of the effective (= approximate ) potential in the MT (= muffin-tin or core ) sphere.
This-p.1-abstract-first says -- Free parameter, No prediction
" error that arises from the linearization in linearized augmented-plane-wave (LAPW) basis functions around
predetermined energies E and show that it can lead to undesirable dependences of the calculated results on method-inherent
parameters such as energy parameters E and muffin-tin sphere radii."
This other website on full-potential DFT also says ↓
p.16(p.14) says -- Fake potential needed
"All-electron methods have to cope with the singularity. Since this singularity cannot be dealt with variationally, one typically works here with basis functions, which are the
numerical solution of (-Δ + Veff-El)φ = 0 of the effective (spherical) potential (= fictitious potential ) containing the singularity, computed in a sphere around the atom at a given energy parameter"
p.22(or p.20)-2nd-paragraph says -- Free energy parameter
"the standard FLAPW (= full-potential LAPW ) functions ul
and dot-u
plus a
further radial function ulo. This new radial function is constructed in the same way as ul,
but with a different energy parameter"
p.29(or p.27)-4.3 artificially choose Fermi energy (= EF which is about valence electron's energy, which is different in different atoms ) and weight parameter ω (= different in different wavenumber or momentum k ). ← No quantum mechanical prediction
p.30(or p.28)-(87) uses this artificially-chosen weight parameter ω to determine electron density in different k or momentum states.
p.32(or p.30)-(99) artificially determines energy parameters El by using this artificially-chosen weight parameter ω (= occupation number f, this p.7 )
↑ As a result, not only in fictional pseudo-potential but also in all-electron full-potential methods, quantum mechanics and its mainstream one-pseudo-electron DFT have to artificially choose various ad-hoc parameters such as energy parameters, weight, muffin-tin core radius, local energy.. which is Not a quantum mechanical prediction.
Ab-initio (constrained-) random phase approximation (= cRPA ) is said to guess Hubbard interaction energy U parameters (= this ad-hoc U energy parameter must be often empirically chosen ).
But even this cRPA is Not true ab-initio, hence, cannot predict any physical values such as Hubbard U Coulomb interaction energy parameters in solids.
The random phase approximation (= RPA ) often uses all-electron (full-potential) linearized augmented plane wave method (= LAPW ) that must artificially choose energy parameters in different bands ( this p.4-right ). ← Not prediction of energies
This cRPA's (fake) prediction of Hubbard U or Coulomb interaction parameters heavily relies on the artificially-chosen parameters such as band energies ( this p.2-3rd-paragraph ), energy window (= choose which energy bands should be included in estimating Hubbard U parameters, this p.6-D, this p.14,p.16-3.5 this-p.3-III,Table.I ), Muffin-tin atomic radius, lattice parameters, basis-set ( Wannier ) wavefunctions ( this p.5-right, this p.5-D. this p.47, this 6~9th-paragraphs ) with No quantum mechanical prediction.
In addition to this artificially-chosen energy parameters, calculated energies and Hubbard U interaction values are affected by various ad-hoc freely-adjustable parameters such as Slater integrals (= F, this 5~7th-paragraphs, this p.8-9, this p.2-right ), double-counting energy correction ( this p.10-2.1, this p.3-left-2~3rd-paragraphs ), and energy frequency (= ω, this p.28,43,69, this p.15, this p.7,p.9 ) in DMFT.
As a result, all quantum mechanical methods are unable to predict any physical values (= so they are fake ab-initio or fake first-principle ) due to their dependence on artificially-chosen fitting parameters.

Feel free to link to this site.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}