Home page
AI is useless for cancers
Today's microscopes
cannot see atoms
(Fig.1) Today's X-ray crystallography, NMR, cryo-electron microcopy are useless, unable to obtain real atomic mechanism or proteins' structures. ← No drug discovery
The current conventional methods such as X-ray crystallography, NMR, cryogenic-electron microscopes (= Cryo-EM ) are useless for drug development, unable to clarify atomic mechanisms of proteins, enzymes.
In the most popular X-ray crystallography, the target proteins must be purified, concentrated and crystallized, and the diffraction or interference patterns of X-ray passing through the crystallized protein are measured to estimate proteins' structures.
↑ The drawback of this X-ray crystallography is that most proteins can Not be crystallized ( this-2nd-paragraph, this-lower-limitation ), and the static protein structures obtained by this X-ray crystallography (= recorded in protein databank or PDB ) are often different from dynamical proteins inside bodies.
So this X-ray crystallography (and AI, Alphafold trained on static proteins of this PDB ) is useless for drug development, unable to clarify atomic mechanism of dynamical biological reactions or diseases.
This 1. crystalline samples says
"The requirement for a crystalline sample is one of the most significant restrictions of x-ray crystallography...
Complex, big, or membrane-embedded proteins frequently cause them to fail"
See this-lower-table-disadvantage.
NMR (= nuclear magnetic resonance spectroscopy irrelevant to quantum mechanics ) measures the nuclear magnetic moment or spin to determine 3-dimensional structures of some small proteins.
NMR can deal with only very small molecules (= clarifying the very important membrane's proteins' structures is impossible ), and often fails to capture the atomic structure when the nuclear magnetic moments are small or samples are aggregated ( this-p.2, this-last-conclusion, this-middle-disadvantage of NMR ).
The 5~6th paragraphs of this site say
"large and dynamic biomolecular complexes, membrane proteins, and macromolecular assemblies that have traditionally been challenging for other structural biology techniques like X-Ray crystallography or nuclear magnetic resonance (NMR) spectroscopy."
This-middle-Disadvantages of NMR spectroscopy says
"The application of NMR in large biomolecule analysis is limited"
"Large amounts of pure samples are needed to achieve an acceptable signal to noise level"
"Highly sensitive to motion. This can lead to signal distortions in artifacts"
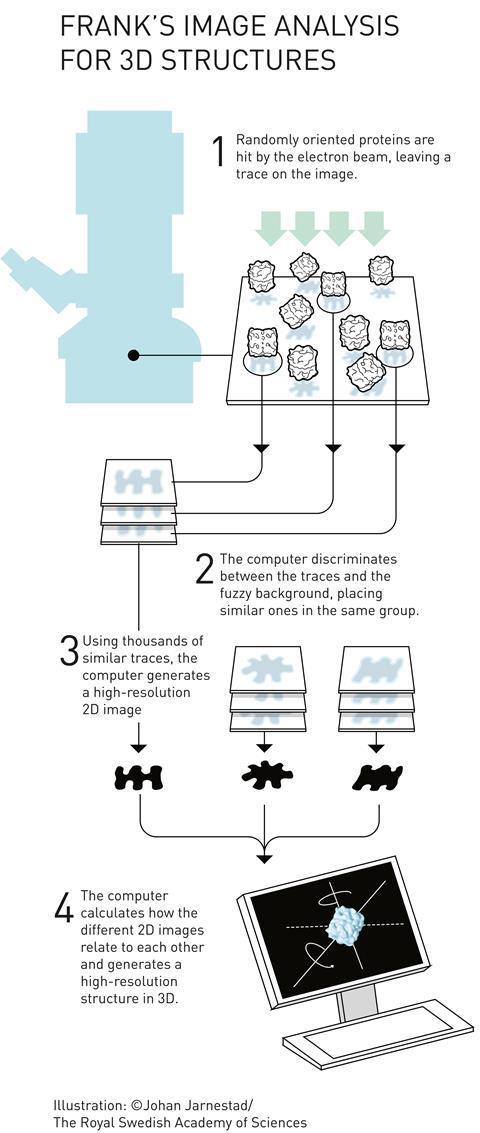
Cryogenic-electron microscopes (= Cryo-EM ) bombard the rapidly-frozen protein samples with high-energy electrons to estimate the target proteins' structures.
The problem is this cryo-EM (= single particle analysis or SPA ) based on detecting randomly-scattered electrons often has bad resolutions (= 3 ~ 4Å ) that can Not distinguish single atoms (= single atomic size is 1Å ).
This-p.11-conclusion (2023) says
"Although the current highest resolution
reached 1.2 Å, most of the reported cryo-EM structures
are still at the 3–4 Å level (= Not atomic level ), and therefore the structural
information on drug molecule binding target sites usually needs to be combined with higher resolution protein
structures obtained from X-ray crystallography"
↑ So cryo-EM unable to know atomic mechanism of proteins is useless for drug development.
This-7th-paragraph (2025) says "However, cryo-EM's resolution typically doesn't match the atomic-level precision of crystallography,"
This-introduction-1st-paragraph (2025) says
"However, building atomic models from cryo-EM density maps remains challenging in many cases."
This-middle-current limitations and challenges in cryo-EM (2025) says
"Membrane proteins can be difficult to purify"
"The high-energy electron beam can damage the sample, limiting the resolution of the reconstruction."
Even the latest Cryo-EM research on this hyped news (8/1/2025) got only about 3Å resolution ( this-p.3, this-p.1 this-p.5-right-1st-paragraph ), which cannot distinguish single atoms (= 1Å, this-p.17 )
Cryo-EM needs to prepare purified homogenous protein samples to estimate the protein structure by averaging many scattered electrons, which is extremely difficult, ( this-key steps in protein sample preparation ), unable to investigate membrane's proteins ( this-3~4th-paragraphs ).
↑ If sample proteins include felexible domains and irrelevant molecules, cryo-em cannot resolve these protein structures ( this-lower-challenges ).
This-6th-paragraph-2.sample heterogeneity says
"the constitutional and conformational differences between molecules need to be minimized. Therefore, the starting point for a successful project requires size-exclusion purified, chemically and structurally homogeneous samples"
This-lower-Challenges and limitations (6/27/2024) say
"The inherent intra-particle flexibility also remains a limiting factor in cryo-EM, which can lead to blurring and difficulty in achieving high-resolution structures. "
Cryo-ET (= cryogenic-electron tomography ) has much worse resolution (= 20~50Å, this-The foundation of Cryo-ET ) that cannot distinguish each single atom, either.
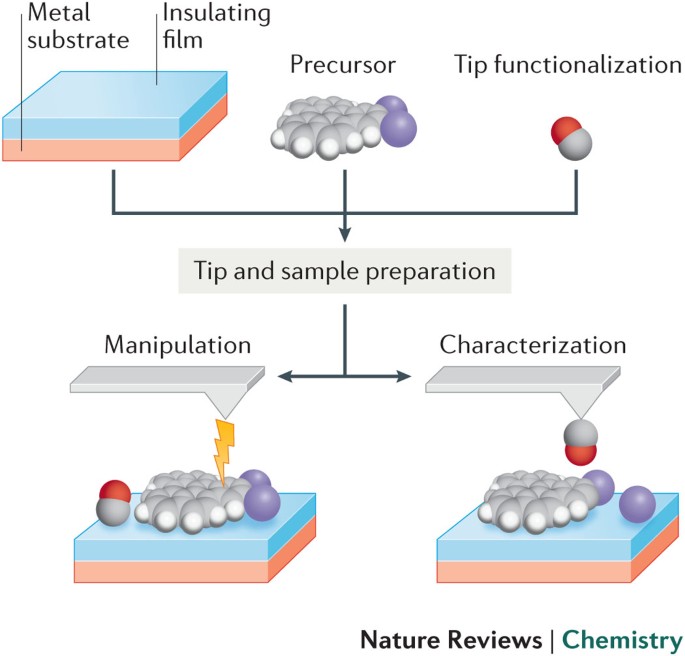
The only way to know the precise atomic mechanism and structures of proteins is to use multi-probe atomic force microscopes directly manipulating single atoms and molecules, which is hampered by the current unphysical quantum mechanical atomic model (= this is why the current AI, Alphafold trained only on bad protein structures is useless, making bad prediction ).
↑ The practical multi-probe atomic force microscopes can directly see and distinguish single atoms of proteins.
↑ Unlike other traditional methods such as cryo-electron microscopy (= bad resolution ), X-ray-crystallography and NMR,
the practical multi-probe atomic force microscope, which can directly observe and touch (= and manipulate ) individual atoms over the target proteins, membranes and cell, does Not need to purify proteins (= which process may change the original proteins' structures ).
If we observe and gradually break the target proteins by artificial molecualr bond breaking, we can know any 3-dimensional and inner atomic structures of proteins by multi-probe atomic force microscopes.
So replacing today's useless single-probe-tip atomic force microscopes by practical multiple probes is necessary and urgent to clarify any proteins' atomic mechanisms and structures.
And the multi-probe atomic force microscopes can develop new effective drugs by directly confirming the biological interactions between the target proteins and the newly-designed drug molecules. → cure diseases

Feel free to link to this site.
{kind=link}
{kind=link}