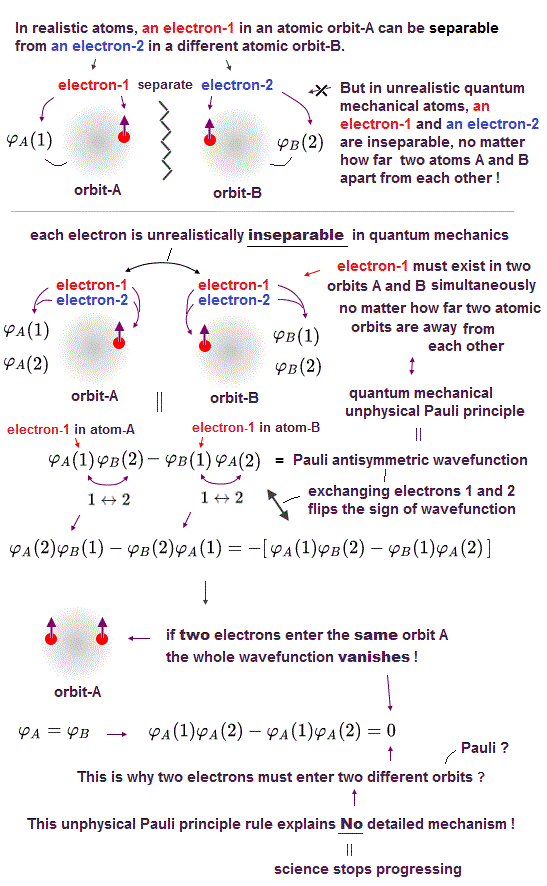
"Truths" have been squashed like the lost integrity in the questionable elections, vaccines, and the current dubious "mainstream science" which stops our advancement forever, and by which the world is gradually regressing to the old bygone systems ?
(Fig.1) Realistic atoms vs. quantum mechanical unreal inseparable atoms
The current mainstream physics stalls our true scientific development by wasting time and money in fictional multiverse, imaginary black holes, bigbang, meaningless gigantic colliders, the paradoxical electron's spin, Einstein relativity, parallel-world quantum computers' hype, spooky superluminal entanglement which cannot send any real information (= hence, impractical forever ), and unseen extra-dimensions which are useless except for selling 'sci-fi' books and political-tool tweeting.
In 1920s, the realistic successful atomic model awarded Nobel prize was unable to handle the complicated three-body problem of helium atom due to the lack of modern computers in 1920s.
Instead, the only remaining unrealistic quantum mechanics had to be accepted reluctantly by the then-physicists as a new (pseudo-)atomic theory despite the fierce opposition even from the current physics founders.
The useless quantum mechanics can Not obtain exact atomic wavefunctions or solve Schrödinger equations in any multi-electron atoms or molecules, it just artificially chooses fake approximate solutions or wavefunctions called trial wavefunctions or basis set in quantum mechanical variational approximate method manipulating many free parameters and integrate it to obtain fake expectation energy value instead of solving the unsolvable multi-electron Schrödinger equations, which quantum mechanical method is just art, not science or ab-initio theory ( this p.6-2nd-paragraph ).
Each atomic orbit is known to have its upper-limit of the number of electrons contained because of some mysterious repulsion originating from Pauli exclusion principle.
Quantum mechanics tries to explain this mysterious Pauli exclusion repulsion using the unrealistic irrelevant spin and unphysical antisymmetric wavefunctions whose sign changes to the opposite when exchanging two electrons (coordinates) 1 ↔ 2 ( this p.11-12 ).
↑ To satisfy this condition of the whole wavefunction's sign change by interchanging any two electrons, in this unphysical quantum mechanical Pauli antisymmetric wavefunctions, every single electron must share all different atomic orbitals simultaneously, which means all atoms sharing a single inseparable electron are unrealistically inseparable !
In this unphysical Pauli antisymmetric wavefunctions, when two electrons try to enter the same orbital with the same (= up-up or down-down ) spins, the whole antisymmetric wavefunctions are supposed to become zero ( this p.11, this p.2 ).
↑ Quantum mechanics and the mathematically-abstract quantum field theory are unable to give any more detailed physical mechanism of how this mysterious Pauli repulsion occurs, instead, they unscientifically claim that the mysterious Pauli repulsion is Not a real repulsive force ( this p.4-lower ) and its repulsive origin is still unknown ( this p.6, this p.3 ). ← Our science progress stops forever due to this unscientific attitude of physicists stopping delving into deeper true mechanisms.
We prove that all quantum mechanical Schrödinger equations for multi-electron atoms and molecules are intrinsically unable to give the exact solutions conserving the constant total energy E in any multi-electron atoms or hydrogen molecule ion, no matter what (fake) approximate solutions or artificial antisymmetric wavefunctions are chosen, so quantum mechanics and its Schrödinger equation are officially proven false, like Erwin Schrödinger himself dismissed his Schrödinger equation causing unphysical dead-alive cats.
This unphysical quantum mechanical Pauli principle's antisymmetric wavefunctions requires every single electron to always exist in all different atoms, nuclei or orbitals simultaneously ( this p.2 ), no matter how far electrons are apart from each other. ← This is impossible.
↑ Because when two atomic orbits with two up and up spins approach each other, and exert mysterious Pauli repulsion to push each other's atom with the same spin's electron away, each electron must already exist in these two different distant atoms or orbitals which are still apart from each other, from the beginning, to satisfy the antisymmetric wavefunction's condition.
↑ These unrealistically inseparable and indistinguishable electrons and atoms required by unphysical quantum mechanical Pauli antisymmetric wavefunction clearly obstruct our atomic nanotechnology by preventing us from using realistic models based on real separable atoms and electrons exerting real attractive and repusive forces originating from some real potential energy sources.
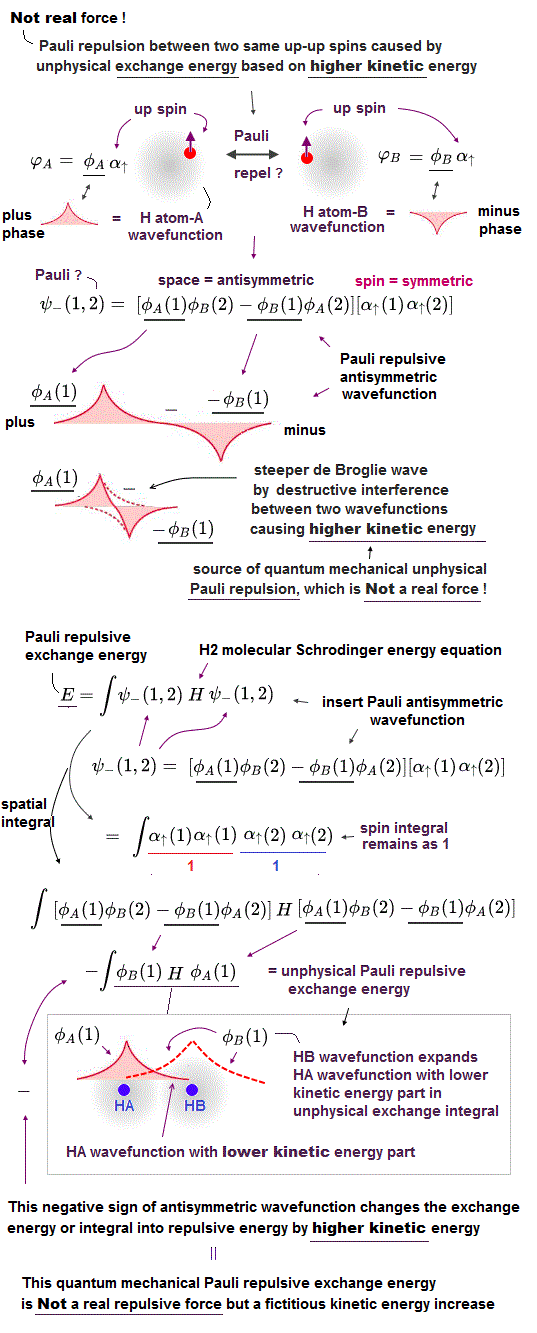
(Fig.2) Origin of quantum mechanical molecular bond is unphysical exchange energy caused by fictitious kinetic energy decrease based on the constructively-interfering symmetric spatial wavefunctions.

In realistic atoms and molecules, when two hydrogen atoms (= H-atom-A and H-atom-B ) approach and form the hydrogen molecular attractive bond, one negative (movable) electron of one H atom has to move and approach the other positive nucleus while the other electron is moving away to the opposite side of the nucleus and avoiding the approaching electron to generate enough Coulomb electric attraction for molecular bonds.
But quantum mechanical electron must always spread all over the place symmetrically around each nucleus as the unphysical electron probability cloud, and these spreading electron clouds cannot approach the other nucleus or avoid the other electron to generate enough Coulomb electric attraction as molecular bond energies.
If the quantum mechanical electron's probability cloud tries to shrink to only one position like a realistically localized electron particle, its contracted and sharpened electron's probability de Broglie wave (= shorter de Broglie wavelength ) increases the electron's momentum or kinetic (total) energies drastically, and cannot obtain the molecular attractive bond energies, which must decrease the total energies.
Hence, quantum mechanics, which cannot rely on real Coulomb electric energies expressed by Coulomb integral, has to rely on the artificially-created unphysical energy called exchange (= resonance ) energies to generate (fictitious) molecular bond energies ( this p.3-4, this p.4-5, this p.4-5-Figure.1, this p.3-last-paragraph, this-lower.Fig.2 ), which are Not real attractive forces ( this last-paragraph ), because these unphysical (attractive) exchange energies are said to be caused by lowering fictitious kinetic energies instead of some realistic potential energy change such as Coulomb electric potential ( this abstract, this p.1-second-paragraph ).
According to the unphysical Pauli antisymmetric wavefunction rule, when two electrons belonging to two different hydrogen (= H ) atoms A and B have the different up and down (= anti-parallel ) spins (= which generate antisymmetric or singlet spin wavefunction ), their spatial wavefunction must take the form of symmetric wavefunction where exchanging two electrons gives the same original equation, hence these two indistinguishable electrons must exist in two different atoms simultaneously to satisfy Pauli (anti)symmetric wavefunctions ( this p.11, this p.4 ).
Pauli antisymmetric wavefunction = spatial symmetric wavefunction × spin-up-down antisymmetric wavefunction → molecular bond attractive exchange energy caused by the spatial symmetric wavefunction ( this p.14-18, this p.4 ).
↑ In this symmetric spatial wavefunction, the hydrogen-A atomic wavefunction with electron-1 (= φA(1) ) and the hydrogen-B atomic wavefunction with electron-1 (= φB(1) ) overlap and interfere with each other constructively (= φA(1) + φB(1) ), which broadens their wavefunctions' de Broglie wavelength and this gentler curvature of de Broglie wavelength allegedly lowers the (fictitious) electron's kinetic and total energies (= bonding, this Figure MO2.2, this p.7-last ).
↑ This fictitious kinetic energy decrease is the origin of quantum mechanical attractive molecular bond exchange energies ( this p.6 ), which are Not real force like Coulomb electric attraction ( this p.5-6 ).
Because the electron's kinetic energy change itself lacking the exchange force or the exchange force's potential energies is Not caused by some real potential energy generating some real force ( this p.8-discussion ). ← kinetic energy is the opposite to the potential energy inside the total energy (= kinetic + potential energies )
Physicists are still unable to give the physical picture or mechanism of this unphysical quantum mechanical exchange energies ( this p.8-lower, this p.11 ).
And the ordinary textbooks deliberately avoid this inconvenient fact that the origin of quantum mechanical molecular bond is Not based on real Coulomb electric force ( this p.3-2nd-paragraph, this p.4-left-2nd-paragraph ).
(Fig.3) Unphysical exchange energy integral consisting of two different HA and HB atomic wavefunctions = ∫ φB H φA

How does quantum mechanical unphysical exchange energy (or integral ) lower the total energy or kinetic energy to generate fictitious molecular bond attractive (exchange) energy ?
Quantum mechanics often chooses fictitious approximate H2 molecular symmetric wavefunction consisting of two hydrogen atomic-A (= HA ) and B (= HB ) 1s wavefunctions ( this p.3-(8) ).
Putting Schrodinger H2 molecular (total) energy equation (= H ), which is the sum of kinetic energy expressed as derivative acting on wavefunctions (= on the right ) and Coulomb potential energy, between these two chosen symmetric wavefunctions, and integrating it ( this p.3 ), this gives two types of integral: one is the ordinary Coulomb energy integral consisting of two same H-atomic wavefunctions, and the other is unphysical exchange energy integral consisting of two different H-atomic-A and B wavefunctions ( this p.11-13 ).
The origin of the unphysical quantum mechanical molecular attractive bond energy is caused by one H atomic-A wavefunction spreading to the other H atomic-B nucleus lowering kinetic energy (= as the H atomic-A electron is farther away from HA atomic nucleus, its kinetic energy is lower ) even while the Coulomb potential energy between the H atomic A's electron and H atomic-B's nucleus is lower.
↑ This quantum mechanical fictitious molecular bond mechanism means the conservation of the constant total energy (= kinetic+Coulomb potential energies ) is violated, because even when Coulomb potential energy between HA-electron and HB-nucleus is lower, HA-electron keeps decreasing its kinetic energy. ← total energy (= lower Coulomb potential + lower kinetic energies ) is decreasing, Not the constant or conserved.
And in the unphysical exchange energy or integral putting Schrodinger total energy equation H between HB atomic wavefunction on the left and HA atomic wavefunction on the right, the other HB atomic higher probability wavefunction around the HB-nucleus expands this lower-kinetic energy region of HA atomic wavefunction near HB atomic nucleus, while the Coulomb potential energy between HA-electron and HB-nucleus is lower near HB-nucleus.
So the quantum mechanical unphysical exchange energy lowers the total energy by lowering kinetic energy (= decrease electron's kinetic energy even in the lower Coulomb potential energy region around the other H atomic nucleus ) by violating the conservation of the constant total energy E.
This means all quantum mechanical molecules based on unphysical exchange energies, which artificially lower electron's kinetic energy even without higher potential energy change or source by violating energy conservation law, are illegitimate and unreal.
(Fig.4) Two wavefunctions with the opposite plus and minus phases destructively interfere and increase the electron's kinetic energy = Pauli repulsion ?
Pauli repulsion or antibonding state is said to be generated between two electrons with the same up-up (or down-down ) spins in two hydrogen atoms HA and HB ( this p.3 ).
This Pauli repulsive antisymmetric wavefunction is the product of the spacial antisymmetric wavefunction, where exchanging two electrons 1 ↔ 2 flips the whole equation, and the same parallel spin symmetric (= triplet ) wavefunction ( this p.16-18, this p.1-2 ).
Pauli repulsive antisymmetric wavefunction = spatial antisymmetric wavefunction (= causing negative repulsive exchange energy or increasing kinetic energy ) × the parallel-same spin symmetric wavefunction.
In this antisymmetric spatial wavefunction between two electrons with the same up-up parallel spins, two H atomic wavefunctions interfere destructively, sharpen their wavefunction's de Broglie wave, and increase the electron's kinetic energy (= resultantly increasing total energy ), which is supposed to generate Pauli repulsive antibonding ( this p.6-2nd-last paragraph, this p.27, this introduction-3rd-paragraph ).
So quantum mechanical Pauli repulsion is Not a real repulsive force, but an result of the magically-increased (fictitious) kinetic energies ( this p.9-10, this p.3-left-middle ). ← This mysterious energy source of the increased kinetic energy or its repulsion-generator potential energy are unknown and lacking in quantum mechanics.
The sign of this Pauli repulsive exchange energy integral is the opposite from the molecular attractive exchange energy integral exploiting the decreased kinetic energy, which also shows Pauli repulsive exchange energy is the result of the illegitimately increased kinetic energy violating total energy conservation law.
(Fig.5) Unphysical quantum mechanical exchange energy's kinetic energy change cannot be the real potential energy causing real forces
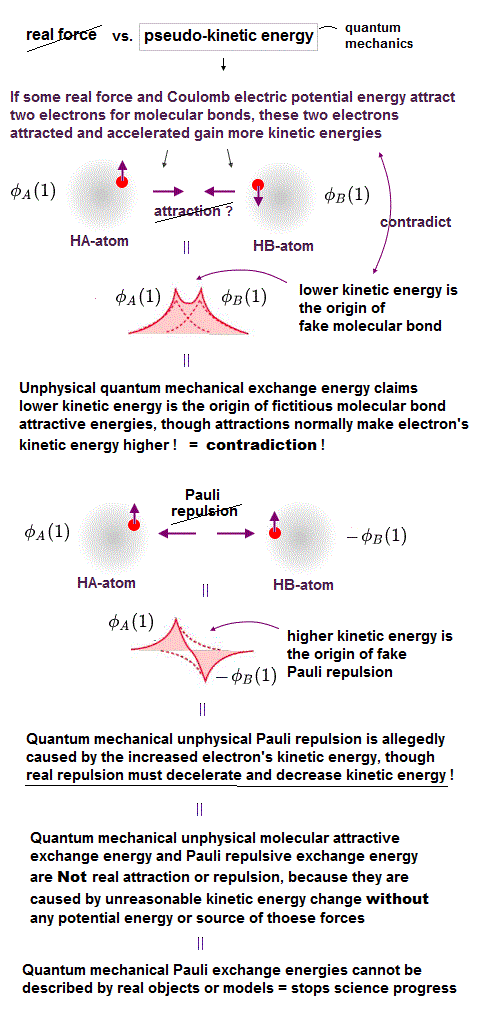
In the normal attractive force such as Coulomb electric attraction between two particles with opposite positive and negative charges, when these charges approach each other, the Coulomb electric attraction between these two charges (= such as a negatively-charged electron and a positively-charged proton ) becomes stronger, and these two charges accelerate and increase their kinetic energies.
But in the unphysical quantum mechanical molecular attractive bond, the opposite and paradoxical thing is said to happen.
When two electrons with the opposite spins in two atoms approach and attract each other to form the molecular bond, these two electrons magically decelerate and lower their kinetic energies !
And in the normal repulsive force such as Coulomb electric repulsion between two particles with same positive (or negative charges ), when these charges approach each other, the Coulomb electric repulsion between these two charges becomes stronger, and these two charges decelerate and decrease their kinetic energies.
But again, the unphysical quantum mechanics says the opposite, paradoxical things: when two electrons with the same parallel spins approach each other and Pauli repulsion between them becomes stronger, these two electrons magically accelerate and increase their kinetic energies ( this p.13 2nd-paragraph ) !
↑ This quantum mechanical Pauli repulsion is the opposite to the real repulsive force ( this p.9-10 ), which clearly contradicts the fact that Pauli repulsion can be measured as normal repulsion when you touch some objects.
↑ This contradictory quantum mechanical Pauli exchange energy based on fictitious kinetic energy change clearly obstructs the development of the realistic atomic science based on realistic atomic models and forces.
Because when we measure some Pauli repulsion on some atoms and want to express this actually-measured Pauli repulsion using the realistic potential causing real repulsion, the unreasonable quantum mechanics prevents it by claiming that Pauli repulsion is Not a real force or generated form some real repulsive potential energy source, instead, it is caused by the magical electron kinetic energy increase lacking any real potential energy source of it !
↑ So developing some real atomic model using real forces by setting real Pauli repulsive potential energies or sources around each atom is impossible as long as we adopt the unphysical quantum mechanical Pauli exchange energies based on the magical change of kinetic energy. ← Science and nano-technology stop progressing forever.
(Fig.6) Distinguishing and separating each different atom and electron are indispensable for developting useful nano-technology and nano-machines, which is prevented by the contradictory quantum mechanical Pauli antisymmetric wavefunction or exchange energy which forbids separating different electrons or different atoms.
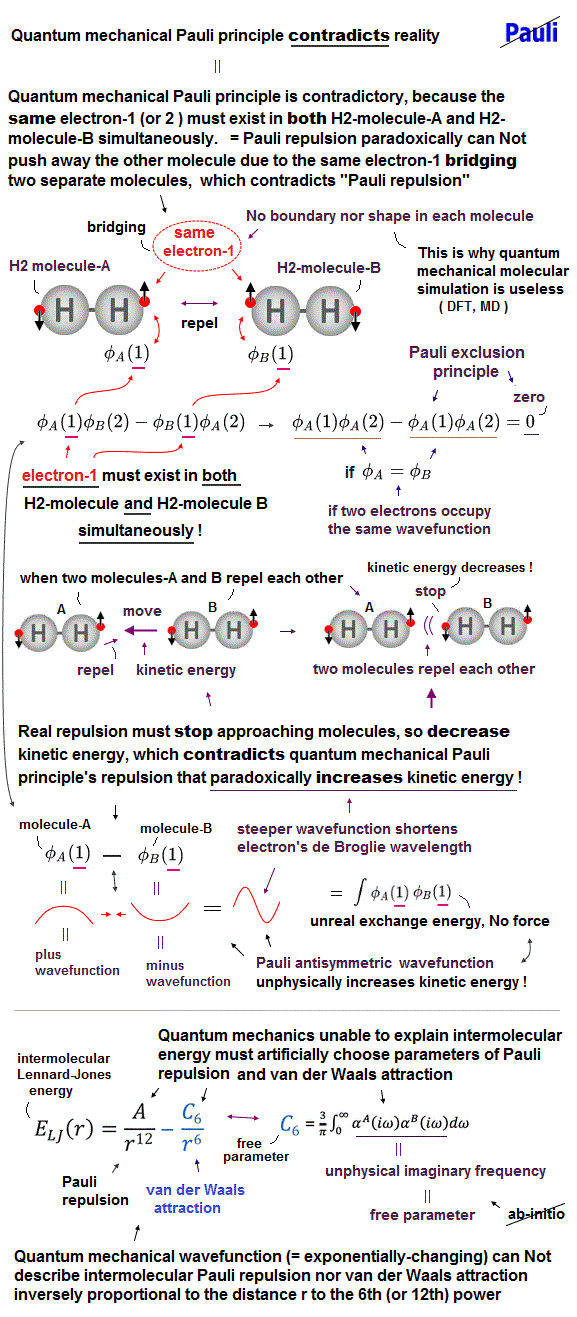
We already have the excellent technology of observing and manipulating each single atom one by one using atomic force or scanning microscopes.
But we still cannot use those excellent technology of manipulating each single atom to build or design some useful nano-machines which have the potential to cure any deadly diseases now. Why ?
In fact, the current nano-technology is great, but the current 100-year-old atomic theory or fantasy quantum mechanics has prevented it from developing for a long time and forever.
To push, pull and move each atom or objects, we need to use some "real forces", separate, distinguish different atoms, measure real contact force or Pauli repulsive force acting on each atom and express these actually-measured atomic forces using some simple realistic atomic model with real potential energies (which mean the concrete shape or size of each atom ) causing real (Pauli repulsive) forces. ← But this realistic setting of real Pauli repulsion is impossible in quantum mechanics !
Pauli repulsion can be measured as "real repulsion" as contact forces also in our daily life, when we touch some solid objects.
Pauli repulsion means when our fingers approach and touch some solid objects, those fingers are stopped by the solid surface or some real repulsive force caused by some real repulsive poential energies giving the shape of the solid object (= the finger's kinetic energy is decreased by being pushed back by the touched solid objects in the case of real repulsive forces ).
But as I said, the unphysical quantum mechanics says the opposite paradoxical things: Pauli repulsion could increase kinetic energy of the finger, when the finger gets closer to the solid surface, which means quantum mechanical Pauli repulsion cannot stop our finger from getting through some solid objects, because our finger accelerates and increases its kinetic energy when it gets closer to the solid surface instead of decelerating and lowering kinetic energy being pushed back by repulsion !
↑ So the unphysical quantum mechanical Pauli principle prevents us from using some realistic atomic model or Pauli repulsive potential energy with no paradoxes causing real repulsion, even when we can measure each precise Pauli repulsion on each atom using the atomic force microscopes !
Furthermore, when our finger approaches and touches some solid objects which stops the finger from penetrating the object, quantum mechanics ridiculously claims that the same electrons of our finger must have already existed also inside the distant solid objects to stop our finger by Pauli repulsion, even when our finger has Not reached or touched the solid objects.
↑ Because the unphysical quantum mechanical Pauli antisymmetric wavefunctions allegedly generaing Pauli repulsive exchange energies require every single electron (inside our finger ) to already exist in all different atoms including the distant objects in advance to exert Pauli repulsion and prevent the finger's atom or electrons from approaching or penetrating the solid object.
↑ If electrons of our fingers do not exist in the distant objects from the beginning, when our finger approaches and touches the solid object, this solid object cannot exert Pauli repulsion or stop our finger using Pauli antisymmetric wavefunctions where even single electron must be indistinguishable and existing in all different atoms simultaneously, because antisymmetric wavefunctions means exchanging any two electrons's positions gives the same wavefunctions (= each electron must exist in all different atoms and positions beforehand ) except for the sign flip.
So as long as we accept this unrealistic quantum mechanical model, we can never distinguish or separate different electrons existing in different atoms, hence, we cannot move different atoms separately (like moving different car parts for constructing a car ) or express real chemical reactions involving electron's transfer from some atoms to different atoms in the realistic way.
As a result, no matter how excellent technology of manipulating each single atom we already have, the unscientific and paradoxical quantum mechanical model prevents us from utilizing those already-existing useful technologies for manipulating atoms and building useful nano-machines !
Quantum mechanics never admits the actually-measured Pauli repulsion is one of real forces, and instead, it makes a ridiculous claim that we cannot touch objects even when our finger actually touches the solid object !
Actually, all the current physicists try to use the quantum mechanical approximate pseudo-model called density functional theory (= DFT ) which unrealistically treats all different electrons in different atoms as one combined big pseudo-electron experiencing some pseudo-potential energies. ← We are Not allowed to treat each different atom or electron separately (= using separate multi-electron coordinates ) in the quantum mechanical unphysical atomic model.
And due to this fictional quantum mechanical model, all physicists can do is artificially create various fictional particle model called quasiparticles with unreal masses and charges to (wrongly) interpret various physical phenomena, also when real atoms or atomic forces are measured by the excellent atomic or scanning microscopes.
↑ This useless quantum mechanical model giving up real particle or force models is a main culprit of forbidding us from advancing useful nano or medical technology for curing deadly diseases.
(Fig.7) If we try to make the three-electron wavefunction mixing the up and down spins, this wavefunction vanishes by canceling the plus and minus terms due to the paradoxical quantum mechanical Pauli antisymmetric rule !

Textbooks often write about two-electron helium and hydrogen molecule, but rarely mention three-electron atoms such as Lithium or molecules.
Because quantum mechanical Pauli principle is unable to treat all atoms and molecules with more than two electrons mixing the up and down or spacial symmetric and antisymmetric wavefunctions ( this p.3-6, or this-(1)-(11) ) !
In the upper figure, the atom-A has an electron with down-spin (= down-spin ↓ is often denoted as β ), the atom-B has an electron with up-spin (= up-spin ↑ is often denoted as α ), and the atom-C has an electron with up-spin.
So the molecular bond attractive exchange energy based on spatial symmetric wavefunctions (= singlet ) between two up-down anti-parallel spins must be generated between two electrons-1 (= down-spin ) and 2 (= up-spin ), and between two electrons-1 (= down-spin ) and 3 (= up-spin ).
See this p.2, this p.1-4, this p.8, this p.22
And the Pauli repulsive exchange energy based on spacial antisymmetric wavefunction (= triplet ) between the same up-up spins must be generated between two electrons-2 (= up-spin ) and 3 (= up-spin ).
↑ These spatial symmetric and antisymmetric wavefunctions have different inconsistent normalization coefficients, so all these separate wavefunctions must be united into one wavefunction mixing spacial symmetric and antisymmetric wavefunction to explain atoms and molecules with more than two electrons, though this is impossible in the quantum mechanical Pauli antisymmetric wavefunctions.
For example, we prepare one term which is the product of three atomic wavefunctions of φA↓(1) (= atomic-A wavefunction with down spin containing the electron-1 ), φB↑(2) (= atomic-B wavefunction with up spin containing the electron-2 ), and φC↑(3) (= atomic-C wavefunction with up spin containing the electron-3 ) = φA↓(1)φB↑(2)φC↑(3).
When exchanging two electrons 1 and 2, this term's sign should remain unchanged as "positive", and this term's electrons' labels change into φA↓(2)φB↑(1)φC↑(3), because the electron-1 exists in the atomic-A wavefunction with down spin and the electron-2 exists in the atomic-B wavefunction with up spin, which must form the spatial symmetric wavefunction with respect to interchanging two electrons 1 and 2 in these terms.
Next we exchange the electron 2 and 3 in this second term of φA↓(2)φB↑(1)φC↑(3), which changes into φA↓(3)φB↑(1)φC↑(2)
↑ No sign change of these spatial symmetric wavefunctions, because the electrons-2 and 3 exist in the atomic-A with down spin and the atomic-C with up-spin. = the up-down spin wavefunction must be symmetric which doesn't change the sign under the exchange of two electrons.
In the same way, when we exchange two electrons 2 and 3 in the first term of φA↓(1)φB↑(2)φC↑(3), this term flips its sign like -φA↓(1)φB↑(3)φC↑(2).
↑ Because the electrons-2 and 3 exist in the atomic-B wavefunction with up-spin and the atomic-C wavefunction with the same parallel up-spin, hence, these two electrons with the same up-up spin must form the spacial antisymmetric wavefunction which must change its sign under the exchange of two electrons' coordinates or labels.
And if we exchange two electrons 1 and 3 in this term of -φA↓(1)φB↑(3)φC↑(2), this does not change the sign like -φA↓(3)φB↑(1)φC↑(2).
↑ because the electrons 1 and 3 exist in the atomic-A with down spin and the atomic-B with up spin, which must form the spatial symmetric wavefunction in the term of -φA↓(1)φB↑(3)φC↑(2).
As a result, this three-electron molecule needs the contradictory terms of plus (= φA↓(3)φB↑(1)φC↑(2) ) and minus ( -φA↓(3)φB↑(1)φC↑(2) ) signs, which cancel each other !
↑ This result shows the quantum mechanical Pauli principle cannot be applied to the case of three electrons or more than two electrons mixing up and down spins.
Only the spatial wavefunction (= not spin part ) is important, contributing to the total energy, but the contradictory quantum mechanical singlet-triplet rule cannot obtain the legitimate spacial wavefunction for three-electron Lithium separated from spin part ( this p.12-third-paragarph, this middle symmetry of three-electron wave functions = this case is self-contradictory where only one spin-up-down bond causes molecular attractive bond exchange energy, and the other spin-up-down bond causes only Pauli repulsive exchange energy ).
For example, the three-electron Lithium (Hylleraas) wavefunction ( this p.3-(6) ) must contain the paradoxical terms like the exchanging the 1st and 3rd electrons paradoxically changes the sign φ(1,2,3) → -φ(3,2,1) or does Not change the sign -φ(2,3,1) → -φ(1,3,2), which is inconsistent with respect to symmetric or antisymmetric wavefunctions under exchanging two electrons, and disagrees with the original Pauli antisymmetric rule.
↑ In true wavefunctions (= if they existed ), wavefunctions of different electrons (= 1,2,3..) must be inseparable, which must also contain terms and variables like r12 (= distance between electron-1 and electron-2 whose wavefunctions are inseparable ) as shown in Hylleraas wavefunctions of helium ( this p.4-6 ).
↑ But in atoms (= such as Lithium ) or molecules with more than two electrons, it is impossible to get true (inseparable mixed) wavefunctions that must contain symmetric (= between two different spins ) and antisymmetric (= between same spins ), hence, quantum mechanical wavefunctions are proved to be wrong.
(Fig.8) Integral of the different up-down spins vanish ! ← any exchange energies between two electrons with the different up-down spins, which are the sources of quantum mechanical molecular bond attractive energies, must become zero. ← No molecular bond in quantum mechanics !

Quantum mechanical Pauli principle cannot describe atoms or molecules with more than two electrons mixing up and down spins, which means quantum mechanics cannot describe the multi-electron wavefunction mixing spacial symmetric wavefunction (= causing molecular bond attractive exchange energy between up-down singlet spins, this p.3 ) and antisymmetric wavefunction (= causing Pauli repulsive antibond exchange energy between up-uo or down-down triplet spins ).
To deal with this serious paradox, quantum mechanics artificially changed the rule, and started to say all multi-electron atomic and molecular wavefunctions must be expressed only as (nonphysical Pauli) antisymmetric wavefunctions (= No symmetric spacial wavefunctions ) or Slater determinant ( this p.1-5 ) where No molecular bonds expressed as attractive symmetric exchange energies (or No bonding exchange integral ) can be generated when atoms or molecules contain more than two electrons. ← No molecular bonds ! It means quantum mechanics is false.
In quantum mechanics, all wavefunctions must be expressed as Pauli antisymmetric wavefunctions or Slater determinants ( this p.7-8 ) where each atomic wavefunction consists of spatial wavefunction and spin wavefunction parts (= α↑ is up-spin, and β↓ is down-spin ).
In exchange integrals of two different atomic wavefunctions, the spin integral of two different up and down spins always becomes zero (= ∫αβ = 0 = No molecular bond attractive exchange energy between the different up-down spins ), and only the spin-integral of two same up-up or down-down spins remains as non-zero (= only Pauli repulsive antibonds between the same up-up or down-down spins remain ) in atoms or molecules with more than two electrons.
↑ It means in quantum mechanics, No molecular attractive exchange energy integral between two different up-down spins is generated (= No molecular bonds in quantum mechanics ! ), and only unnecessary Pauli repulsive exchange energy between the same up-up or down-down spins remains ( this p.5-6, this p.37, this p.10-lower ).
This is a fatal flaw of quantum mechanical Pauli principle based on unphysical antisymmetric wavefunctions.
(Fig.8') ↓ Singlet-triplet theory is contradictory in Pauli principle of more than two electrons, so quantum mechanics is false.

In two-electron helium atom, quantum mechanics insists there is difference between the energy levels (= ex. 1s2p of helium ) of singlet (= 1P ) expressed as spin-up ↑ (= α ) and spin-down ↓ (= β ) and triplet (= 3P = spin-up-up, spin-up-down, spin-down-down symmetric ) wavefunctions ( this p.9 ).
But as shown in this, this quantum mechanical singlet-triplet theory is invalid in more than two electrons such as lithium.
So quantum mechanics illegitimately changed the originial Pauli principle definition based on the helium-singlet-triplet states into generalized atomic Pauli principle version expressed as Slater determinant or Pauli antisymmetric wavefunction where each wavefunction consists of spatial (= φ ) and spin (= α-up-spin, β-down-spin ) parts (= which contains No spatial symmetric wavefunction even in spin-up-down ↑↓ case ).
The problem is this generalized Pauli principle antisymmetric wavefunction or Slater determinant can Not distinguish singlet and triplet energy levels. = There is only one form of antisymmetric wavefunction (= two terms connected by minus ) with two electrons of spin-up and down, as shown in the above figure.
So in all atoms with more than two electrons, the original singlet-triplet theory ( this p.8 ) is invalid, which is inconsistent with the quantum mechanical claim that even magnesium and calcium with more than two electrons should have singlet and triplet energy levels.
This clear self-contradiction shows quantum mechanics is wrong.
In boron with three valence electrons, the 2s-2p2 state splits into five different energy levels (= 4P + 2D ), while in aluminum that also has three valence electrons, the same type 3s-3p2 state splits into only three energy levels (= 4P ), which is inconsistent (= Aluminum paradoxically ingnores 3p2 singlet inside 3s-3p2 state ). ← Singlet-triplet based on the original Pauli principle becomes invalid in three valence electrons.
As shown in beryllium with two valence electrons, if 2s2p triplet state (= 3P ) splits into three total angular momentom states (= J=0,1,2 ).
In the 2p2p (or 3p3p ) states, when two electrons enter two different p orbitals whose angular momentums are perpendicular to each other, the total angular momentum in one direction should reflect only one of those two perpendicular p orbitals, hence, it should split into only two (= J = 1/2 or 3/2 ) which contradicts magnesium 3p2 splitting into three
states (= J = 0,1,2 )
Furthermore, helium's three energy levels of 1s2p triplet (= 3P = 2,1,0 ) are inconsistent with beryllium's (= 2s2p ) and magnesium's (= 3s3p ) triplet states' order 3P = 0,1,2. ← No legitimate consistent explanation in quantum mechanics.
Beryllium (= Be ) and singly-ionized boron (= B+ ) have exactly the same structure with two valence electrons, so these two atoms should have the same-order energy levels.
But the orders of 2s3s and 2p2 between Be and B+ are completely different and inconsistent.
In carbon with four valence electrons, 2s2-2p2 splits into five states (= probably they assume three triplets + two singlets in 2p2 inside 2s2-2p2, which are also contradictory, because this does not include the total angular momentum J = 1 in spin-up-down in two perpendicular 2p orbitals ) while the same type 2s2-2p3p paradoxically splits into ten states (= only this includes two types of triplets ), though 2s2-2p2 and 2s2-2p3p should have the same types of triplet.
In neon with eight valence electrons where they probably try to treat the 6 electrons as one independent singlet state with spin-zero, then, they should split the remaining 2p-3s (of 2p53s ) into three triplet + one singlet like helium and beryllium, but they split 2p53s into two pairs irrelevant to the original triplet-singlet theory, which is inconsistent.
(Fig.9) Molecular orbital (= MO ) theory requires each single electron to exist in multiple different atoms simultaneously, which also causes unrealistically-inseparable electrons or atoms, and MO cannot avoid unnecessary Pauli repulsive exchange energy, so false.
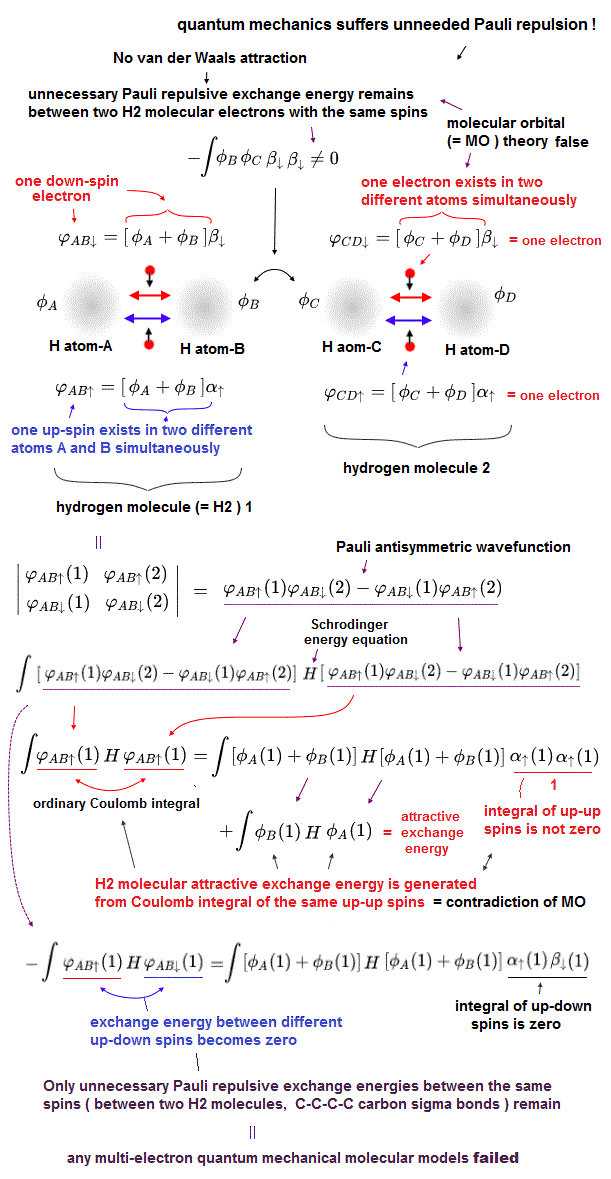
Quantum mechanical Pauli principle expressed as antisymmetric wavefunction or Slater determinants have fatal flaws, and it can Not describe any molecular bond attractive exchange energies between the different up-down spins, and only unnecessary Pauli repulsive exchange energies between the same up-up or down-down spins remain.
To handle this quantum mechanical contradiction, the molecular orbital theory (= MO ) was invented, though this ad-hoc molecular orbital theory cannot eliminate unnecessary Pauli repulsion after all, either.
In this ad-hoc molecular orbital theory, each single electron must always spread over more than one atoms or orbitals = each electron's molecualr orbital always becomes a linear combination of multiple atomic wavefunctions ( this p.7(or p.5), this p.4, this p.3 ). ← This unrealistic condition is necessary for generating the fictitious molecular bond attractive energies in MO by avoiding the vanishing attractive exchange energies between the up-down spins.
It means electrons of molecular orbital theory are unrealistically inseparable into different atoms due to their constantly-spreading molecular orbitals, and the ad-hoc molecular orbitals cause non-existent ionization states ( this p.27, this p.14-15-(11)-(13) ), hence, the molecular orbital theory often failed to explain experimental results ( this p.25 ).
This molecular orbital wavefunction also has to be expressed as the unphysical Pauli antisymmetric wavefunction or Slater determinant consisting of multiple molecular orbitals ( this p.14, this-(9.6.1)-(9.6.2) ) where each orbit consists of a spatial molecualr orbital (= linear combination of multiple atomic orbitals ) and spin part.
↑ This means the molecular orbital theory also causes unnecessary Pauli repulsive exchange energy between two molecular orbitals with the same spins, which means any quantum mechanical multi-electron models failed to incorporate Pauli principle expressed as the antisymmetric wavefunctions or Slater determinants.
To avoid this unnecessary Pauli repulsive exchange energies, all molecular orbitals must be "orthogonal" to each other, which means all exchange energies or overlap integral between any two different molecular orbitals must be zero ( this p.6, this p.3 ).
↑ When any exchange energies (= or overlap integral ) between two different orbital wavefunctions become zero due to their orthogonal wavefunction's relationship regardless of their spin states, neither molecular bond attractive exchange energies nor annoying unneeded Pauli repulsive exchange energies seem to appear in the energy calculations. ← But even these artificial "orthogonal" wavefunctions cannot avoid unnecessary Pauli repulsive energies after all, so the molecular orbital theory is wrong, as I explain later.
If all exchange energy integrals become zero due to artificially choosing the orthogonal molecular orbital wavefunctions, how does the ad-hoc molecular orbital theory generate the molecular attractive (or Pauli repulsive ) exchange energies, though the exchange energy is zero ?
As shown in the upper figure, even if exchange energies or exchange integrals are zero due to the artificially-chosen orthoginal wavefunctions, the normal Coulomb integral parts (= the product of two same wavefunctions with the same spin ) automatically contain the molecular attractive exchange energies caused by each molecular orbital which is a linear combination of multiple atomic orbitals, which is the trick of molecular orbital theory to generate molecular attractive exchange energies.
Each molecular orbital = ψ = φA(1) + φB(1), hence, the normal Coulomb integral of ∫ |ψ|2 = ∫ | φA(1) |2 + | φB(1) |2 + ∫ 2 φA(1)φB(1) ← the attractive exchange energy +∫φA(1)φB(1) is automatically contained in the normal Coulomb integral of each single molecular orbital.
The problem is that it is basically impossible to make all exchange energies between all different combinations of different molecular orbitals zero or orthogonal, especially in the spherical s atomic orbital like in hydrogen 1s atomic wavefunctions (= exchange integral of two different hydrogen atomic 1s orbitals cannot be zero or orthogonal ).
Molecular orbital theory tries to make artificial orthogonal wavefunctions combining plus (= bondong, this p.68 ) and minus (= antibonding ) phases of atomic wavefunctions ( this p.1-(2) ), but the orthogonal wavefunctions or zero exchange energies mean the overlap region between two orbitals are artificially removed (= which equals the sharpened de Brogloe wave increasing Pauli repulsive kinetic energies, this p.49-51 ), hence, the artificially-created orthogonal wavefunctions also include unnecessary Pauli repulsive exchange energies in their Coulomb integral, after all.
So the ad-hoc molecular orbital theory cannot handle intermolecular van der Waals attraction or molecular sigma bonds such as C-C-C-C multiple carbon bonds due to its inability to eliminate unnecessary Pauli repulsion especially between two atomic spherical s orbitals.
Physicists gave up the theoretical prediction based on this falied molecular orbital theory or multi-electron quantum mechanical model, and instead, invented the empirical Huckel method which can treat only π bonds at first (= because p orbital originally consists of plus and minus phases, and their π bond molecular orbitals can easily become orthogonal ) by artificially adjusting free semi-empirical energy parameters obtained from experiments ( this p.4 ), which cannot predict any molecular enregies or treat spin exchange energies ( this p.2-2nd-last paragraph ).
As shown here, quantum mechanical Pauli principle expressed as unphysical antisymmetric wavefunctions or Slater determinants intrinsically contradict multi-electron atomic or molecular wavefunctions.
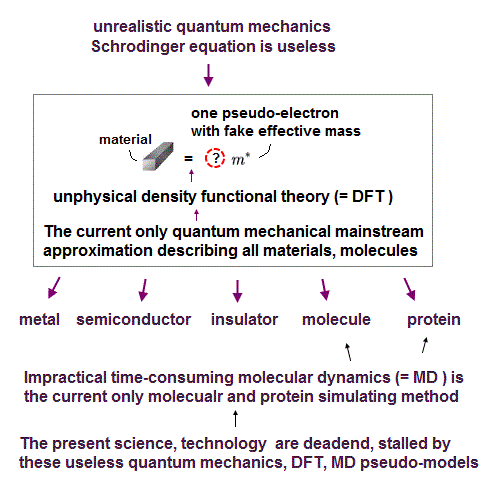
This is why only unrealistic one-pseudo-electron approximation called density functional theory (= DFT ) remains as the most-widely used quantum mechanical calculation tool, though this DFT also failed to incorporate Pauli principle.
(Fig.10) DFT replacing the whole many-electron material by only one pseudo-electron model with artificially-chosen pseudo-potential is not only unable to explain Pauli principle but also obstructing the real atomic science advancement forever.
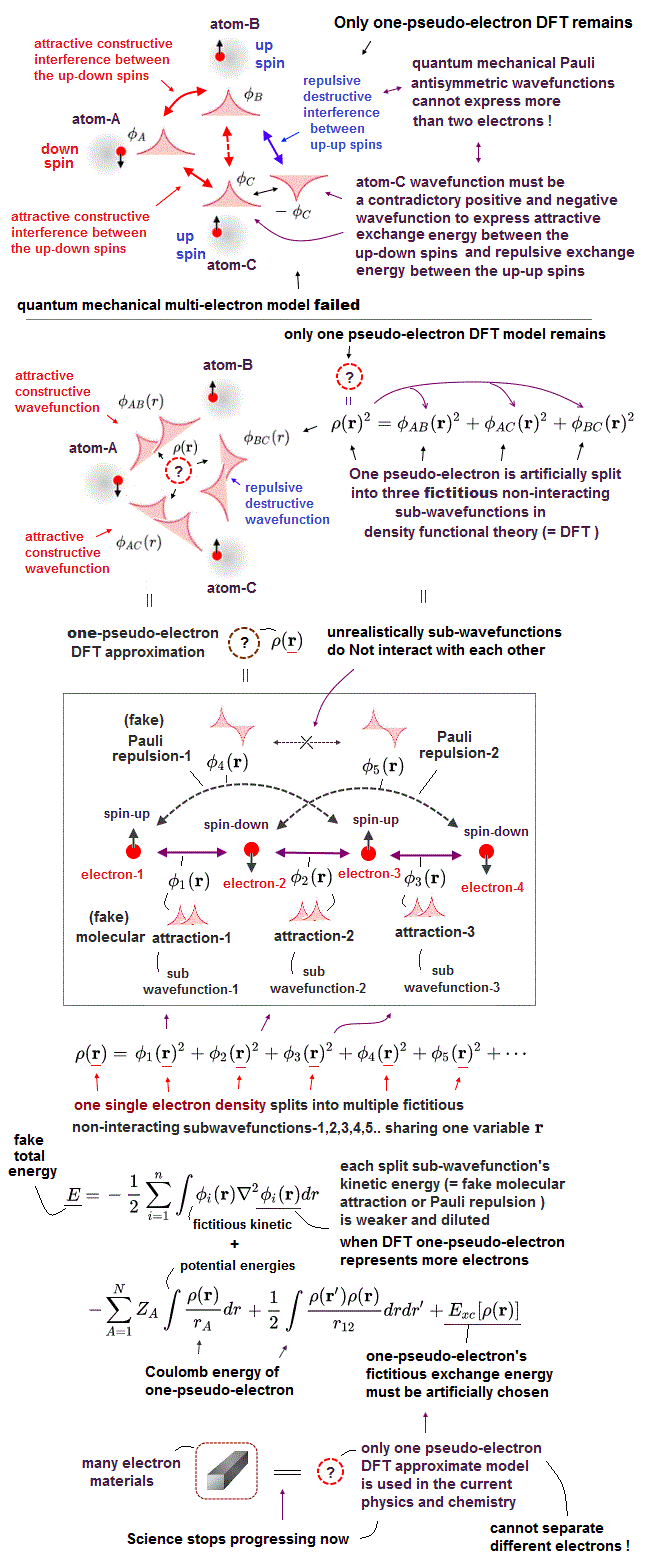
After all the ad-hoc multi-electron quantum mechanical molecular models failed to incorporate Pauli principle expressed as antisymmetric wavefunction, almost all physicists started to rely on the unphysical density functional theory (= DFT ) or Kohn-Sham theory which unrealistically treats all different electrons and atoms inside many-electron materials as only one inseparable pseudo-electron approximate model using only one electron's coordinate (= r ) in almost all fields of physics and chemistry in vain ( this p.3-5, this p.6-upper ).
As I said, this unrealistically-inseparable electron of quantum mechanics and its most-widely-used approximate DFT model (= often combined with impractical pseudo-classical molecular dynamics ) are the main culprit of stopping our basic and applied science progress forever.
First of all, this DFT using only one pseudo-electron or one electron's variable (= only one electron's coordinate r ) in any complicated multi-electron molecules cannot describe the original multi-electron Pauli antisymmetric wavefunction which becomes zero when it contains only one electron's variable.
Pauli antisymmetric wavefunction = φA(1)φB(2) - φB(1)φA(2) → φA(1)φB(1) - φB(1)φA(1) = 0, when this contains only one electron-1 like one-pseudo-electron DFT where the original Pauli principle's antisymmetric wavefuntion does Not work. ← The current mainstream quantum mechanical DFT is self-contradictory and wrong.
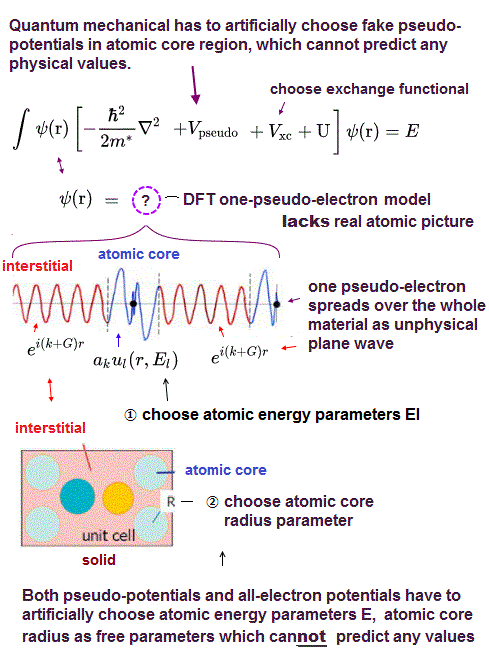
DFT changes multi-electron potential energies into fictitious pseudo-potential energy based on only one pseudo-electron called exchange-correlation functional potential energy, whose exact universal potential energy form is unkown ( this p.27 ), and physicists must artificially invent and choose this DFT fictitious exchange-correlation potential functional ( this p.6 ).
↑ Artificially-choosing pseudo-potential functional energy and one-pseudo-electron's self-interaction error mean DFT itself is unable to predict any atomic properties (= DFT tries to obtain deceptive relative energy due to its inability to get true absolute atomic energy ) = DFT is Not an ab-initio theory at all ( this p.10-second-paragraph ).
Of course, this one-pseudo-electron approximate DFT often failed to explain the actual multi-electron phenomena such as intermolecular energies ( this p.18-low ), metals ( this p.16-right ) and large molecules ( this p.2 ) due to lack of the universally-exact exchange-correlation pseudo-potential functional ( this p.17, this p.17-20, this p.13-upper, this p.2-first-paragraph ).
↑ Even the current most-widely-used emprical DFT hybrid exchange functional called B3LYP ( this p.8-1.5.4 ) often failed to give exact energies ( this p.3 ).
DFT has to artificially split one-pseudo-electron into multiple fictitious non-interacting subwavefunctions sharing only one electron's coordinate = r (= No physical meaning, this p.15 ) to mimic the ordinary molecular attractive constructively-interfering symmetric wavefunctions and Pauli repulsive destructively-interfering wavefunctions ( this p.12, this p.6, this p.34, this p.7 ).
If these fictitious DFT subwavefunctions can interact or interfere with each other like multi-electron wavefunctions, it causes paradoxical situation where some subwavefunctions cancel each other and vanish when there are more than two electrons.
As shown in the upper picture of Fig.10, when atom-A has an electron with spin-down, atom-B has an electron with spin-up. and atom-C has an electron with spin-up, all these three atoms-A,B and C must have the same positive-phase atomic wavefunctions to cause the constructive interference lowering kinetic energies or the molecular bond attractive exchange energies between the down-spin electron in atom-A and the up-spin electrons in atom-B and C.
But the Pauli repulsive antibond destructively-interfering exchange energy must be generated between two same up-up spins of atom-B and C, which means the atom-C's electron wavefunction must flip its phase to negative sign (= φC → -φC ), so the resultant positive and negative atom-C's wavefunctions cancel each other, and we cannot express the molecule consisting of three atoms with one down-spin and two up-spins, if these three wavefunctions are interacting (= not non-interacting ) with each other.
The fictitious non-interacting subwavefunctions cause serious problem in DFT's kinetic energy, which disproves the validity of DFT itself.
As shown in this, when there are only two electron (of two atoms ), only one molecular bond is formed between these two atoms, and all two electrons' density can concentrate on this single molecular bond formation like two hydrogen atomic wavefunctions are used for only one hydrogen molecular bond formation.
But when there are six electrons (or six atoms ), there are as many as 15 interatomic interactions between different combinations of these six electrons, hence, we have to split six electrons' charge density into 15 independent non-interacting subwavefunctions describing molecular attractive or Pauli repulsive exchange energies acting between these six electrons.
↑ So when there are only two electrons with two atoms, the single molecular bond between these two atoms can be strong enough by using all these two electrons' charges for its single molecular bond (= 2/1 ), but when there are six electrons, these six electrons' charges must split into 6/15 smaller charge for each interatomic interaction, which becomes far weaker than the case of two electrons.
↑ This means these DFT fictitious non-interacting subwavefunctions cannot generate enough interatomic exchange energies, when there are many electrons in the system considered, because the limited number of electrons' density must be divided into many noninteracting independent subwavefunctions for all possible different interatomic interactions among many electrons.
So after all. one-pseudo-DFT approximation also failed to express the quantum mechanical Pauli principle, which basically has the unavoidable fatal flaw especially in more than two electrons.
But this failed one-pseudo-electron DFT is the current only usable quantum mechanical approximate method even after physicists observe actual multiple atomic behavior using the atomic force microscopes ( this last ). Physicists often use various artificially-created pseudo-potential functionals and adjustable parameters for this useless DFT model ( this p.2-left, this p.5-right ).
Physicists artificially created various interatomic pseudo-potential energies such as Lennard-Jones potential containing empiriically-fitted parameters where Pauli repulsion cannot be obtained from the quantum mechanical theory ( this p.3-left-2nd-paragraph ).
As a result, all the ad-hoc quantum mechanical models such as valence bond, molecular orbital, and one-pseudo-electron DFT are unable to explain its unphysical Pauli principle's exchange energies, which fatal contradiction proves the quantum mechanics is definitely wrong.
Physicists often use the pseudo-classical approximate model called molecular mechanics or molecular dynamics in larger molecules or proteins.
The point is this pseudo-classical molecular mechanics or dynamics is not ab-initio but just one of empirical theories with No power to predict anything.
This molecular mechanics or dynamics based on experimentally-adjusted free parameters or pseudo-potential energy called force field, does Not contain any real electrons at all
So these pseudo-classical molecular mechanics (= MM ) or dynamis with No real electrons cannot describe any chemical realctions such as bond breaking and change involving real electron's transfer ( this p.2-last ).
In conclusion, unless we accept the real atomic model using actually-moving electrons and real de Broglie wave, our basic atomic science would never advance, as shown in the paradoxical quantum mechanical Pauli exchange energies lacking any real forces.
(Fig.11) Four Helium atoms (= A,B,C,D ) are arranged tetrahedrally. ← Each unphysical molecular orbital ( in four He1, He2, He3, He4 molecular orbitals ) contains two electrons up and down spins: each electron must be unrealistically spreading all over four Helium atoms.

Here we show one typical example where the unphysical quantum mechanical molecular orbitals can neither remove unnecessary Pauli repulsion nor generate the necessary van der Waals attraction between four helium atoms (nor generate covalent sigma bond's energy in molecules with multiple atoms, this p.1-left-2nd-paragraph ), hence, quantum mechanics is inconsistent with the experimental results, and wrong.
In the upper figure, four helium atoms (= each of He-A, He-B, He-C, He-D atoms contains each helium 1s atomic orbital wavefunction φA, φB, φC, φD, respectively ) are arranged regular tetrahedrally (= distances between any two helium atoms are supposed to be equal, meaining all the overlap or exchange integrals (= S ) between any two of these helium atoms are also equal ).
Each atomic orbital contains one electron, which condition is called "normalization" expressed as the integral of two same atomic wavefunctions being 1 like
∫φAφA = ∫φBφB = ∫φCφC = ∫φDφD = 1
And the overlap (= or exchange ) integral between any two different helium atomic orbital wavefunctions becomes S due to the same interatomic distance between any two electrons ( this-(7) ) like
∫φAφB = ∫φAφC = ∫φAφD = ∫φBφC = ∫φBφD = ∫φCφD = S.
Four helium atoms contain the total eight electrons = four electrons with up spins and four electrons with down spins.
Due to the fatal flaws of quantum mechanical Pauli antisymmetric wavefunctions, any molecular bond attractive exchange energies between two different up and down spins become zero, and only unnecessary Pauli repulsive exchange energies between two electrons with the same up-up or down-down spins remain.
↑ To avoid this quantum catastrophe, quantum mechanical ad-hoc molecular orbital theory demands that any overlap or exchange integrals between any two different molecular orbitals must be zero, which artificial condition is called "orthogonal" or "orthonormal (= orthogonal + normalization, this p.14, this p.6 )."
Each molecular orbital can contain the maximum two electrons with up and down spins due to Pauli exclusion principle, so four different molecular orbitals (= He1, He2, He3, He4 ) are necessary to describe interactions between four helium atoms containing eight electrons (= two electrons with up and down spins × four molecular orbitals ).
Quantum mechanical molecular orbital is unrealistic, because each electron must always spread all over four helium atoms, and each electron's unphysical molecular orbital (= MO ) must be expressed as the linear combination of multiple (= four helium ) atomic orbitals (= AO, this middle-methane-case ).
Each molecular orbital contains one electrons, so the ( Coulomb ) integral of the same two molecular orbital (= total probability density of each electron ) must be 1, like
∫He1He1 = ∫He2He2 = ∫He3He3 = ∫He4He4 = 1
which normalization condition gives different normalization coefficients to different molecular orbitals.
As shown in the upper figure, we can find four (unphysical) molecular orbitals which are orthogonal to each other, which means the overlap integrals of any two different molecular orbitals must be zero like
∫He1He2 = ∫He1He3 = ∫He1He4 = ∫He2He3 = ∫He2He4 =
∫He3He4 = 0.
↑ But even if this artificial orthogonal molecular orbitals are created, quantum mechanical molecules cannot avoid unnecessary Pauli repulsive exchange energies between the same spins (= between four electrons with the same up spin of four helium atoms, and between four electrons with the same down spin of four helium atoms ), after all, so quantum mechanics is false.
Due to the orthogonal molecular orbitals (= any overlap or exchange integrals between any two different molecular orbitals are zero ), only ordinary Coulomb integrals (= integral of two same molecular orbitals ) remain, when we integrate the chosen wavefunctions with Schrodinger energy equation (= H ) to obtain (fake) total energy.
↑ But each molecular orbital is the linear combination of multiple different atomic orbitals, so the ordinary Coulomb integrals of the same molecular orbitals or the square (= probability ) of each molecular orbital automatically contain the exchange energy integrals.
The nomalization coefficients of Pauli repulsive antibond molecular orbital including the minus-phase atomic orbital tend to be bigger than the attractive molecular bond orbital including only the positive-phase (spatial) atomic orbitals.
So after all, the sum of the ordinary Coulomb integrals of four molecular orbitals gives the Pauli repulsive exchange energy integrals (= negative overlap or exchange integral = -S ) between the above four helium atoms.
It means the quantum mechnaical molecular orbitals can Not avoid unnecessary Pauli repulsive exchange energies, and cannot express the actual van der Waals attraction between helium atoms without adding artificial pseudo-potential energies. ← Quantum mechanics is proven wrong.
As I said above, quantum mechanical Pauli antisymmetric wavefunctions cannot give van der Waals attraction due to their unnecessary Pauli repulsion.
So quantum mechanics had to artificially make up unphysical pseudo-potential energy called London dispersion energy to express this intermolecular van der Waals attraction ( this p.1-2 ).
This ad-hoc quantum mechanical intermolecular attractive pseudo-potential energy (= dispersion energy ) must artificially adjust parameters such as fictitious imaginary frequencies ω ( this p.13-18, p.22-24, this p.2-5 ) of each molecular (unobservable) instantaneous dipole polarizations and pick up the excited-energy basis set (fake) wavefunctions ( this p.1-4, this p.9-15, this p.1-4 ), empirically ( this p.4-left-last-paragraph ).
Quantum mechanics can explain neither the intermomecular van der Waals attraction energy inversely proportional to the sixth power of the intermolecular distance nor Pauli repulsion inversely proportional to the twelfth power of intermolecular distance.
↑ The intermolecular energies obtained by the original quantum mechanical wavefunctions (= expressed as the exponential function ) must be changed in the exponential way (= e-r/r ) which disagrees with both actual van der Waals attraction (= change obeying -1/r6 where r is intermolecular distance ) and Pauli repulsion (= change obeying +1/r12 ). ← Furthermore strong Pauli repulsive exchange energy always surpasses or dominates over the weaker Coulomb attractive energy in any intermolecular distances (= No van der Waals attraction ! ) based on the quantum mechanical wavefunctions as shown in always-positive antibond (= Pauli repulsive ) energy.
So they tried to deliberately divide the total energy (= Hamiltonian energy H or E ) of two molecules (= A and B ) into the two independent molecules (= against the original molecular orbital theory that must connect all molecules and atoms ) and the independent intermolecular energy (= parameter × V ) that allegedly includes the above van der Waals attractive pseudo-potential energy (= London dispersion = irrelevant to quantum mechanics ! ) as the second-order perturbation ( this p.1-2, this p.3-right-p.16 ), and Pauli exchange repulsion energy.
↑ Both the forms of van der Waals attractive energy and Pauli repulsive energy must be artificially created irrelevant to the original quantum mechanical wavefunctions, and their parameters must be artificially adjusted, in order to fit the experimental intermolecular energies. ← Quantum mechanics failed to explain actual experimental results.
↑ Even this quantum mechanical artificial intermolecular pseudo-energy can Not explain the intermolecular strong Pauli repulsion that must change more rapidly and acutely than the quantum mechanical wavefunction's exponential change in the short-distance ( this p.2-right ).
Some ad-hoc models try to use the exponential change for intermolecular Pauli repulsion, these models also disagree with the experimental observations (= slow-exponentially-changing Pauli repulsion becoming far weaker than van der Waals attraction that becomes stronger obeying -1/r6 in much shorter distance clearly disagrees with the actual intermolecular interaction, this p.4-second-paragraph ) even after artificially adjusting their empirical parameters ( this p.2-left-first-paragraph ).
So after all, for the intermolecular interaction, the failed quantum mechanics has to rely on the (fictitious classical) Lennard-Jones potential energy whose repulsive and attractive energy parameters have to be artificially adjusted irrelevant to the original quantum mechanical theory ( this p.2-right-last ).
The coupled cluster (= CC ) approximate method is called "gold standard ( this p.24 )", but this CC (or CCSD ) also has to rely on artificially-created pseudo-potential as van der Waals energy proportional to 1/r6 (= which cannot be expressed by quantum mechanical wavefunctions + normal Schrodinger equation ), and CCSD is known to be illegitimate, Non-variational method which can often give wrong energy values lower than true energis ( this p.1-left-1st-paragraph, this 1st-paragraph ), depending on artificially-chosen basis set functions.
So quantum mechanics unable to predict any molecular energies is a total failue.
(Fig.12) The most-widely-used quantum mechanical DFT approximation tries to add artificially-chosen pseudo-potential or fictitious exchange energy consisting of only one pseudo-electron (= ρ(r) ) to seemingly fix the unnecessary Pauli repulsive energies caused by multi-electron molecular orbitals in vain.

All muti-electron quantum mechanical theories such as molecular orbitals failed to eliminate unnecessary Pauli repulsive exchange energies.
So physicists had No choice but to create the unrealistic quantum mechanical approximation called density functional theory (= DFT ) or Kohn-Sham theory which unreasonably replaced the whole many-electron material or molecule by just one pseudo-electron model (= unreal quasiparticle model, this p.3, this p.11-top ), which is allegedly moving in some unknown fictitious potential energies called exchange-correlation functionals ( this p.27 ).
Quantum mechanical Pauli exclusion principle must be expressed as unphysical antisymmetric wavefunctions or Slater determinants where exchanging any two electrons should give the same original wavefunction (= so each electron is indistinguishable and inseparable from other different electrons in different positions ) except for the sign flip.
↑ This quantum mechanical Pauli antisymmetric wavefunction or Slater determinant becomes canceled to be zero, when it contains only one electron like
antisymmetric wavefunction = φA(1)φB(2) - φA(2)φB(1) → φA(1)φB(1) - φA(1)φB(1) = 0 (= when it contains only one electron 1 = 2 ).
DFT uses only one pseudo-electron, it means if we apply this one-pseudo-electron of DFT to the multi-electron quantum mechanical Pauli antisymmetric wavefunctions, these wavefunctions vanish !
But even this one pseudo-electron DFT approximation is said to satisfy the same condition as the multi-electron quantum mechanical (useless) Pauli antisymmetric wavefunction or Slater determinants ( this p.2-3rd-paragraph ).
How does DFT using only one electron incorporate the multi-electron quantum mechanical Pauli principle without canceling its antisymmetric wavefunctions ?
As I said, the multi-electron quantum mechanical molecular orbital wavefunctions must be orthogonal to each other ( this p.1-lower ), which means any overlap or exchange (energy) integrals of any two different molecular orbitals must be zero ( this p.14 ), and only the sum of the Coulomb integrals of the same two wavefunctions or the square of the same wavefunctions remains as the total energy.
DFT artificially splits its one pseudo-electron (= ρ(r) ) into multiple fictitious non-interacting sub-wavefunctions squared (= all these split sub-wavefunctions keep sharing only one electron's coordinate r, this-3.2.1, this p.9 ) like
ρ(r) = φ1(r)2 + φ2(r)2 + φ3(r)2 + φ4(r)2
↑ These DFT fictitious artificially-chosen sub-wavefunctions sharing one electron coordinate correspond to the Coulomb integrals of the multi-electron quantum mechanical molecular orbitals.
In quantum mechanical unphysical Pauli antisymmetric wavefunctions, all electrons are unrealistically indistinguishable and inseparable (= as if all multiple different electrons hehave like one pseudo-electron ), so if their molecular orbitals are orthogonal, and only Coulomb integrals remain, it could be changed into one-pseudo-electron DFT model ( this p.30(or p.18)-footnote ).
Actually, these DFT fictitious sub-wavefunctions must be also orthogonal or orthonomal (= overlap or exchange energy integrals of two different sub-wavefunctions must become zero ) to each other ( this p.17, this p.10, this p.8-lower, this p.2-right-last ) like
∫φ1(r)φ2(r) = ∫φ1(r)φ3(r) = .. = 0
↑ It means these artificially-chosen DFT subwavefunctions must be the artificially-constructed orthogonal linear combinations of multiple atomic orbitals ( this p.3, this p.38, this-(1), this p.4-upper ) and always spread over all atoms (= hence, each electron and atom are unrealistically inseparable from other atomic electrons also in DFT ) like the ad-hoc molecular orbitals, as shown in the above case of the four helium atoms.
For example, as shown in the above figure, one DFT subwavefunction φ1(r) and another subwavefunction φ2(r) cancel a part of each other's terms, if DFT's one pseudo-electron can interact with each other like
φ1(r) + φ2(r) = ( φA + φB + φC + φD ) + ( φA + φB - φC - φD ) = 2( φA + φB ) ?
↑ This is why DFT fictitious electron must be non-interacting.
Non-interacting (= independent ) subwavefunctions mean, as the number of electrons in the system considered is larger, the number of interactions among those more electrons is larger, and they have to split the limited number of electron's density into more smaller independent interactions, so each interelectronic or interatomic interaction is weaker, as the electrons increase in DFT, which also disproves DFT and molecular orbital theory.
And these artificial orthogonal DFT subwavefunctions also contain unnecessary Pauli repulsive exchange energies inside their Coulomb integrals of orthogonal fictitious subwavefunctions (= ∫φ1(r)φ1(r) + φ2(r)φ2(r) + φ3(r)φ3(r) + φ4(r)φ4(r) contains unneeded Pauli repulsive exchange energies ) like the multi-electron molecular orbitals.
To eliminate these unnecessary Pauli repulsive exchange energies inherent in the orthogonal (sub-)wavefunctions, DFT rely on various artificially-created pseudo-potential energies called exchange-correlation functionals, which exact form is unknown or non-existent ( this 5th-paragraph, this p.20-lower, this p.6, this p.17 ).
These DFT pseudo-potential energies or exchange-correlation functionals basically consist only of one pseudo-electron density (= ρ(r) or n(r), this p.7-8, this p.7, this p.6,10~ ), hence, these ad-hoc DFT pseudo-potential energies cannot distinguish or separate each different electron or atom. ← DFT is inapplicable to actual nanotechnology which needs manipulation of different atoms and electrons separately.
Inside these DFT pseudo-potential energies or exchange functionals, all different electrons with spins must be treated as one big inseparable pseudo-electron (= DFT has No potential energies separating or distinguishing each different electron with differen spin, though distinguishing each different electron with different spin is indispensable to correctly estimate the attraction between different spins or repulsion between the same spins ).
Hence, these fictitious DFT exchange potential energies cannot be used to push, pull or separate different electrons or atoms. ← Quantum mechanical most-widely-used DFT approximation is completely useless and unable to describe actual multi-electron atomic bahaviors whose knowledge is necessary for developing useful nano-technology.
Furthermore, the quantum mechanical molecular method of artificially choosing either attraction expressed as spacial symmetric wavefunctions or Pauli repulsion expressed as antisymmetric wavefunctions is inherently unable to describe actual intermolecular interactions mixing van der Waals attraction and Pauli repulsion (= creating a mixed symmetric and antisymmetric spacial wavefunction is impossible ).
This is why DFT can Not explain intermolecular van der Waals interactions ( this p.1-failure, this p.3, this p.48, this p.63 ).
DFT tries to artificially create and add various ad-hoc pseudo-potential energies or exchange energy functionals by manipulating many free empirical parameters to explain van der Waals force in vain ( this p.3-4, this p.2, this p.2, this p.22 ).
↑ Despite this irreparable contradiction, the current physicists try to find the illusory universal DFT fictitious exchange potential energy functional consisting of only one inseparable pseudo-electron model, which should paradoxically describe any behaviors of arbitrary numbers of separable differnet electrons in vain.
↑ Finding this dreamlike still-unknown universal DFT pseudo-potential functional allegedly describing any multi-electron behaviors using only one-pseudo-electron model is definitely impossible ( this p.39-last-paragraph ).
As I said, each ad-hoc orthogonal subwavefunction must always spread all over different atoms, which also cannot separate different electrons or atoms inside multi-electron or multi-atomic materials.
So it is intrinsically impossible for the one-pseudo-electron DFT model to distinguish and separate different electrons (= if all electrons are unrealistically indistinguishable and inseparable, all atoms are also inseparable ), which means it is impossible to apply this one pseudo-electron DFT model to the actual multi-electron or multi-atomic materials or molecules.
But due to the quantum mechanical fatal flaws = unphysical Pauli antisymmetric wavefunctions, only one pseudo-electron DFT approximation with artificially-adjustable pseudo-potential energies remain as the calculation tool of (fake) molecular energies in all the current physics and chemistry.
So all the current atomic and applied science miserably stops progressing by these unrealistic and paradoxical quantum mechanical models such as the most-widely-used one inseparable pseudo-electron DFT approximation.
(Fig.13) DFT or Kohn-Sham theory has to artificially add various ad-hoc van der Waals attractive pseudo-potential functionals to the original unphysical exchange energy functionals where all electrons must be treated as one fictitious inseparable electron = ρ(r)
The most-widely used quantum mechanical approximation = density functional theory (= DFT, this 4th-paragraph ) or Kohn-Sham model unrealistically treats all different electrons existing in all different atoms as one big pseudo-electron (= ρ(r) or n(r) ) with only one electron's coordinate (= r, this p.31-36, this p.3-5 ).
So this one-pseudo-electron DFT model is intrinsically unable to treat actual multi-electron or multi-atomic bahaviors, hence, completely useless ( this p.2-left-upper ). ← But this useless unrealistic DFT method dominates all the current quantum physics and chemistry, because of no alternative under the unphysical quantum mechanical model.
Intermolecular interactions are known to consist of van der Waals attraction (= London dispersion forces ) allegedly caused by unknown instantaneous fluctuations of electron's charges with imaginary frequencies ( this p.4-right ) at medium distance and Pauli repulsion at short distance ( this p.10-13, this p.1-left ).
One-pseudo-electron DFT, which cannot distinguish or separate different electrons existing in different atoms, has No ability to describe these actual intermolecular van der Waals attractions which vary depending on the distance between two different atoms or electrons ( this p.8-left-lower ).
DFT has the pseudo-potential energies called "exchange-correlation energy functionals" which exact form is unknown, and many artificial exchange-correlation energy functionals, which allegedly contain various molecular bond attractive and Pauli repulsive exchange energies, have been created with No success.
Physicists had to artificially create and add various ad-hoc (semi-empirical) van der Waals attractive (dispersion) energy functionals (= Edisp or Enl or vdW-DF ) to the original artificially-chosen DFT's unphysical Pauli exchange (correlation) energy functionals (= Exc = they created many different versions of exchange correlation functionals such as ELDA, EGGA, EPBE, EPW86.. this p.6, this p.2-right ).
These artificially-created DFT's van der Waals pseudo-potential functionals designed to be inversely proportional to the sixth power of the distance (= RAB6 that cannot be explained by the original quantum mechanical atomic wavefunctions giving the exponentially-changing potential instead of RAB6 ) between two atoms A and B contain various freely-adjustable parameters such as the van der Waals attractive interaction constant (= C6, ) which must be artificially changed differently depending on different combinations of two atoms, and the damping factors (= fdamp ) which parameters also must be artificially adjusted to be weaker as the distance RAB between two atoms A and B gets shorter ( this p.5-left, this p.2, this-5.21-5.26, this p.2-4, this p.6-left ).
These DFT's artificial ad-hoc van der Waals pseudo-potential functionals (= denoted as Edisp or Enl which means nonlocal London dispersion energy ) are intermolecular attractions, so they must be connected to the DFT's original unphysical Pauli repulsive exchange energy functionals (= Exc or EKS-DFT which has various different forms such as LDA, GGA, PBE, PW86 .. exchange functionals, this p.8 ) to explain the actual intermolecular interaction mixing van der Waals attraction and Pauli repulsive exchange energies ( this p.2-3, this p.3-adjustments, this p.8-right-upper ).
Another artificial parameter s6 or sR included in most of the van der Waals functionals must be empirically determined and adjusted depending on what exchange energy functionals (= Exc ) such as LDA, PBE, BLYP.. are used together with van der Waals functionals ( this p.30, this p.4-left-2nd-paragraph, this p.3-right-2nd-paragraph ).
The point is these ad-hoc van der Waals intermolecular attractive functionals (= Edisp or Enl ) try to paradoxically distinguish only different atoms (= Not electrons ) by choosing different van der Waals attractive parameters (= C6 ) sharing only one pseudo-electron density coordinate (= ρ(r) or n(r), this 6th-paragraph ) without distinguishing different electrons existing in different atoms A and B.
As I said, in order to explain quantum mechanical unphysical Pauli repulsion (= which is Not a real force ! this p.5 ) between two different atoms A and B, each electron must unrealistically exist in both different atoms A and B at the same time using the unphysical Pauli antisymmetric wavefunctions or Slater determinants
Hence, all different electrons of quantum mechanics and its most popular one pseudo-electron DFT approximation must always exist in all different atoms simultaneously and inseparably.
Actually, all the artificially-chosen DFT's exchange pseudo-potential energy functionals (= Exc ) such as LDA, GGA, PBE, PW86.. must consist only of one inseparable pseudo-electron (= denoted by ρ(r) or n(r), this p.4, this p.7-8 ) with one electron coordinate (= r ) combined with various freely-adjustable parameters ( this p.3-5, this p.21-25, this p.1-2 ).
As I said, the DFT's one-pseudo-electron (density ) ρ(r) are temporarily split into multiple fictitious non-interacting subwavefunctions ( ρ(r) = φ1(r)2 + φ2(r)2 + φ3(r)2 + φ4(r)2 +.. ) keeing sharing only one electron's coordinate (= r ) to obtain DFT's fictitious kinetic energies.
↑ Each of these DFT's fictitious subwavefunctions must spread all over the atoms sharing the one common electron's coordinate (=r ) to satisfy the quantum mechanical ad-hoc condition of the orthogonal wavefunctions where all overlap intagrals of all different subwavefunctions must be zero.
Some Pauli exchange (correlation) functionals were artificially corrected for long-range intermolecular repulsive interaction, and they also consist only of one pseudo-electron ρ(r) or unphysical Hartree-Fock exchange integrals of its subwavefunctions φ(r) spreading all over different atoms simultaneously, hence, all the DFT's unphysical Pauli exchange energy functionals are unable to separate or distinguish different electrons existing in different atoms at all ( this p.9-14 ), which is useless for applied science.
As a result, the quantum mechanical unphysical Pauli exchange energies and its most popular approximation DFT's exchange energy functionals consisting of only one pseudo-electron (= ρ(r) ) are unable to describe actual important enzymatic reactions such as atomic bond breaking and rebinding which need the transfer of each different electron from one atom to another different atom, because each quantum mechanical electron must always exist in all different atoms simultaneously to satisfy the unphysical Pauli antisymmetric wavefunction rule.
Under this quantum mechanical unrealistic inseparable pseudo-electron model, it is impossible to predict in which direction each atom will move, when this atom is pushed or pulled by different atoms.
Because when one atom-A pushes (or pulls ) another atom-B, the electrons and nucleus of this atom-A exert some (real) force on another atom-B's electrons and nucleus. ← Unreasonable quantum mechanics does Not allow even this realistic situation where different separable electrons existing in different atoms A and B exert real attractive or repulsive forces on each other due to its unphysical Pauli exchange energies lacking real exchange force.
So in this useless quantum mechanical model, physicists have to artificially choose completely different and irrelevant fake approximate wavefunctions or pseudo-potential energies for different electrons' configurations or different situations from scratch without being able to express real forces and without predicting in which direction each separable atom or electron will be moved by real forces from other atoms or electrons.
As a result, the unphysical quantum mechanics and its most-widely-used one-pseudo-electron DFT are completely useless and inapplicable to actual useful atomic nanotechnology, which is why our basic and applied scientific progress completely stops by unphysical quantum mechanical model now.
To advance useful science such as effective drug discovery, the first thing to do is when we measure each atomic force, size using the current excellent technology such as atomic and scanning microscopes, we need to express those actually-measured atomic ( attractive and Pauli repulsive ) real forces and atomic sizes using real tangible objects or real atomic models with real separable electrons, nuclei and real force carriers such as the real de Broglie wave's (destructive) interference (= origin of real Pauli repulsion unlike the paradoxical quantum mechanical pseudo-kinetic energy increase ).
(Fig.14) Quantum mechanical materials, metals are unrealistic consisting of the unphysically spreading useless wave instead of real separable electrons.
Quantum mechanical most-widely-used one-pseudo-electron DFT or density functional theory is known to be unable to explain ordinary metals, semiconductors, strongly-correlation systems, band energy gap, and excited energies.
Also in these ordinary materials such as metals and semiconductors, the unphysical quantum mechanics and its leading model = DFT have to unscientifically treat all different electrons belonging to different atoms as one big inseparable pseudo-electron model (= denoted as ρ(r) or n(r) ) containing only one pseudo-electron's coordinate r.
So all quantum mechanical pseudo-models such as one-pseudo-electron DFT or Kohrn-Sham equation are unable to describe actual electric currents where each separable electron must be actually moving from one atom to another atom inside metals and semiconductors, which means the tired cliche "quantum mechanics was useful for developing modern computers is a total lie.
Actually, all quantum mechanics can do is describe the whole material or metal as an "unrealistic band" or the nonphysical spreading plane wave consisting of one pseudo-electron ( this p.1-(2), this p.24, this p.39 ) or fictitious quasiparticle with unreal effective mass (= which could be negative mass, this p.6, this-Table1 ) without giving any realistic pictures of individual separable electrons.
Due to unphysical Pauli antisymmetric wavefunction causing fictitious exchange energies, all quantum mechanical models cannot handle or distribute actually separable electrons into different atoms.
↑ It means quantum mechanics cannot describe the realistic situation like an electron-1 existing in an atom-A, while another electron-2 exists in an atom-B, instead, quantum mechanics says an electron-1 (or electron-2 ) must always exist in both different atoms-A and B simultaneously and unrealistically, which means we cannot separate an atom-A containing an electron-1 from an atom-B containing an electron-2, because the same single electron-1 (or electron-2 ) must always bridge these two atoms-A and B simultaneously.
Also in one-pseudo-electron DFT model, each electron must always spread over all atoms in the whole material as one unrealistically-inseparable fictitious electron (= ρ(r) or n(r) ) inside (pseudo-)potential energies and exchange-correlation energy functionals (= Exc ), which exact form is unknown (and non-existent ), but all the current physicists are pursuing it in vain.
In metals and semiconductors, DFT must artificially add another ad-hoc pseudo-potential (= another exchange and Coulomb) energy including freely-adjustable parameter U (= DFT+U ) to the original exchange-correlation energy functionals such as LDA,GGA,PBE.. (→ LDA+U, GGA+U..) in order to artificially deal with the material's strong Coulomb force effect ( this p.3 ).
↑ This means finding DFT's (non-existent and illusory) universal exchange-correlation energy functional allegedly applicable to all metals (= + U ) and non-metals (= U is unnecessary ) is impossible forever ( this p.7, this 7-6th-last paragraphs, this 2nd-paragraph ).
↑ These ad-hoc parameters U must be artificially changed and adjusted into different values in different materials or metals based on experimental results, and quantum mechanics has No power to predict all these energy parameters ( this p.1-left, this p.4-last-paragraph, this p.1-left-last-paragraph, this p.3 ).
Or physicists have to artificially choose (unreal) pseudo-potential energy ( this p.9-left-2nd-paragraph ) or additional perturbation energy (= λV, λ is freely-chosen parameter ) to manipulate total energy where there is No original quantum mechanical prediction ( this p.3-left-lower ).
The most serious irreparable problem of quantum mechanics and its leading model = one-pseudo-electron DFT approximation is they cannot separate different atoms or electrons, hence, their unphysical quantum models cannot use "real (measurable) forces" between atoms to know in which direction each atom will be moved by other external atomic forces, because of No real force allowed in Pauli exchange energies.
In metals, semiconductors and periodic crystals, DFT often aritificially chooses the abstract unphysical plane wave model spreading over all atoms with No real electron's shape (= expressed just as nonphysical spreading functions of eikr or simple sinusoidal curve, this p.16-33, this p.6-12 ) as one pseudo-electron wavefunction or basis set whose kinetic energies (= contained in the de Broglie wavelength ) or the upper energy's cut-off must be artificially adjusted ( this p.22,33, this p.6-right, this p.27-37 ).
↑ These very old quantum mechanical abstract (augmented) plane wave (= APW ) function allegedly representing the (pseudo-)electron model has been amazingly unchanged (= stopped progressing ) and used even now ( this p.9-left ), since it was first invented by Slater in 1937 ( this p.3-47 ). ← This proves our atomic science has made No progress for a long time.
↑ So this plane-wave-shaped electron unrealistically spreading over all different atoms inside materials cannot be divided into different individual electrons belonging to different atoms, which quantum mechanical model cannot be utilized for any practical purpose which needs to move each different atoms (= containing different separable electrons ) separately.
Each electron in quantum mechanical material or metal is often expressed as the (artificially-chosen) spreading plane wave function in the interstitial region (= all place except for near each atomic core ) + atomic (sphere) wavefunction only around each atomic core, which ad-hoc separation methods or parameters must be artificially determined differently in each different materials using abstract nonphysical models, various pseudo-potentials ( this p.11-12, this p.68-69, this p.9 ) and unreal electron's effective masses ( this p.3,14 ).
All these unphysical pseudo-electron's plane wave or DFT's fictitious non-interacting sub-wavefunctions must obey the unscientific quantum mechanical rule = orthogonal wavefunctions where any overlap integrals of two different plane waves must be zero. ← This quantum mechanical meaningless restriction is one of main reasons why quantum mechanical electron wave functions have remained impractical and unchanged for a long time.
As I said, for atoms and electrons to be used for really useful purposes, each different atom with concrete shape (= given by different electrons existing in different atoms ) must be moved separately from other atoms using "real forces (= instead of unphysical exchange energies lacking real physical forces )", which means electrons in one atom also must be separable from other electrons in other atoms, which basic separation is impossible in the unphysical quantum mechanical Pauli antisymmetric wavefunctions or exchange energies.
So in any molecules and materials such as metals, semiconductors and insulators, quantum mechanics is intrinsically unable to describe actually-separable and movable electrons, hence completely useless, and preventing our scientific development forever.
Actually, even in the latest nano-technology research ( Dec.21 2022 ), physicists just manipulated only a single molecule using the scanning microscope, and Never tried to move forward to manipulating multiple molecules or constructing useful molecular nano-machines, because they always and blindly rely on the nonphysical one-pseudo-electron DFT exchange energy functional with the nonphysically-spreading inseparable electron's plane wave model, with No progress, forever ( this p.7-DFT calculation ).

2022/12/19 updated. Feel free to link to this site.
{kind=link}
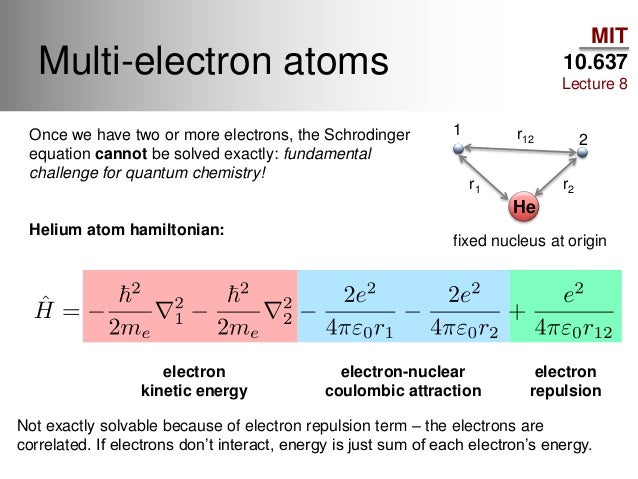{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
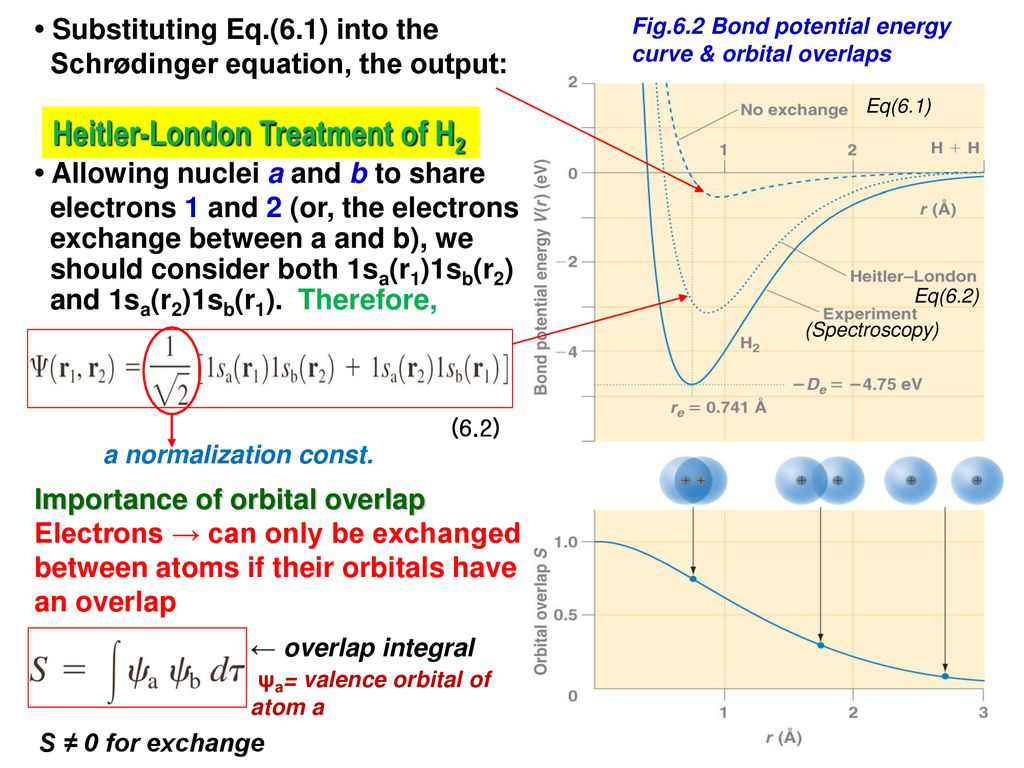{kind=link}
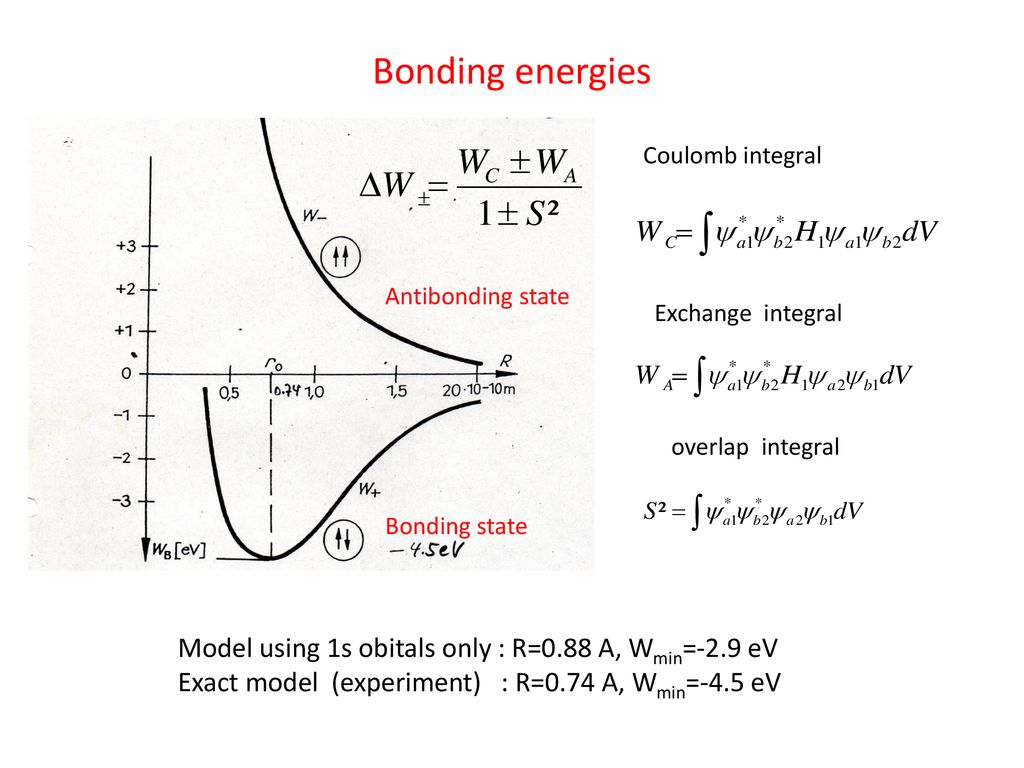{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
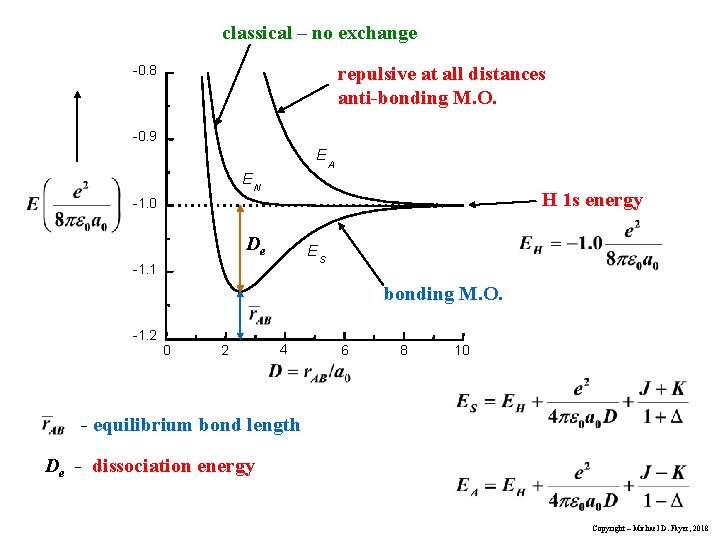{kind=link}
{kind=link}
{kind=link}
{kind=link}
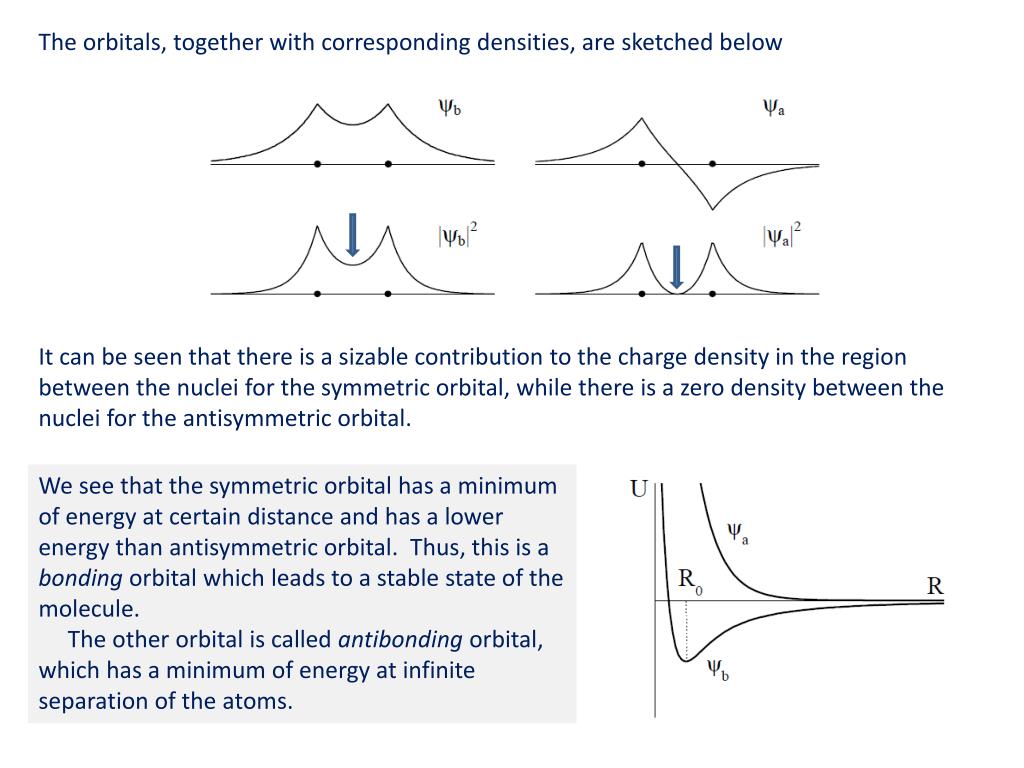{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
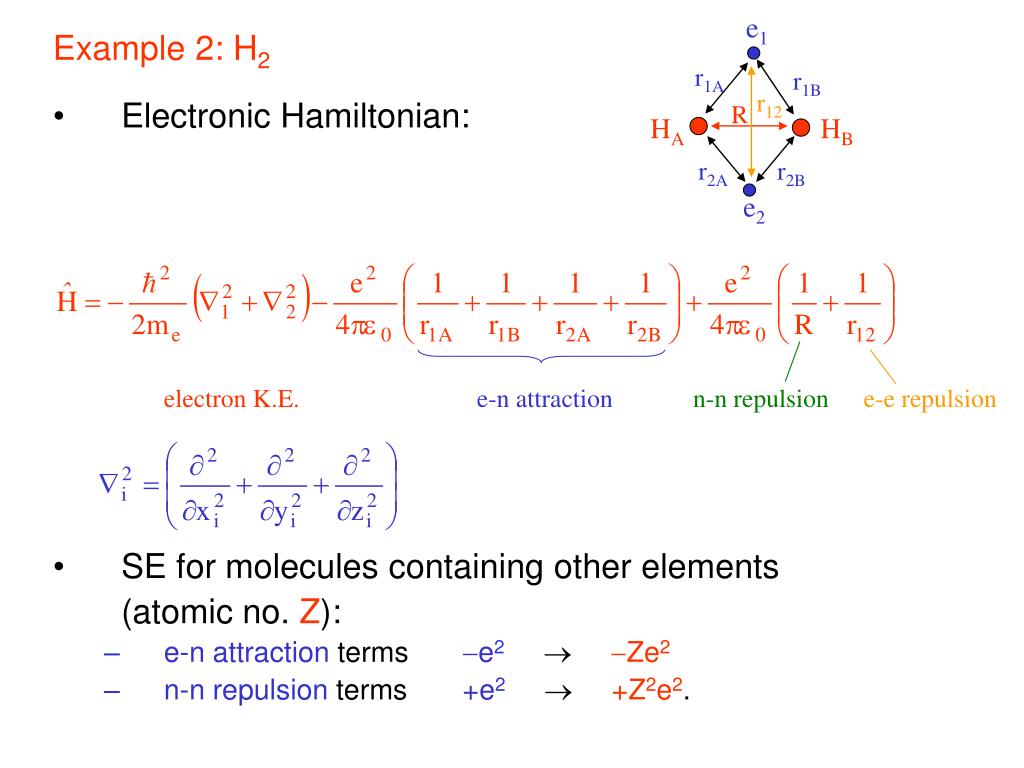{kind=link}
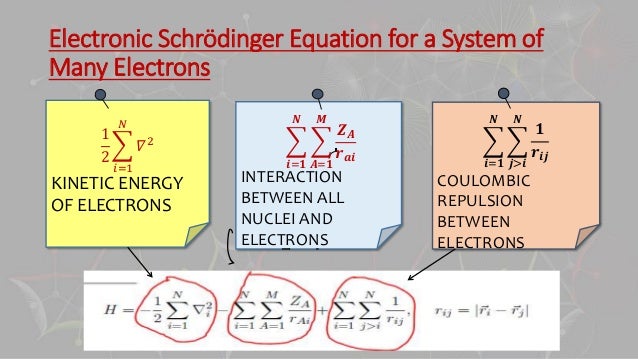{kind=link}
{kind=link}
{kind=link}
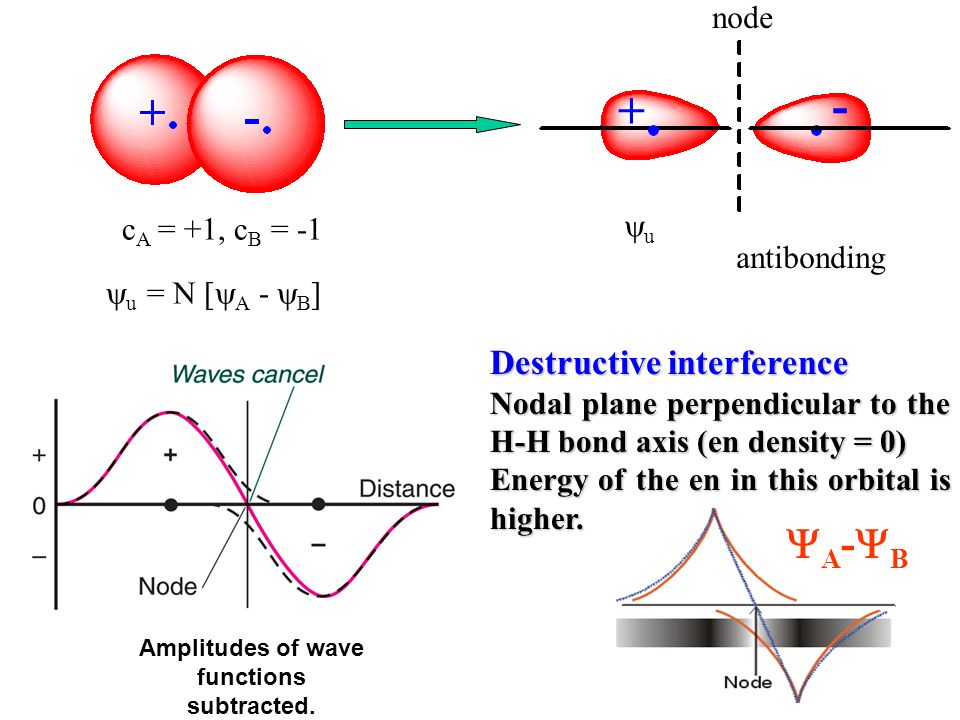{kind=link}
{kind=link}
{kind=link}
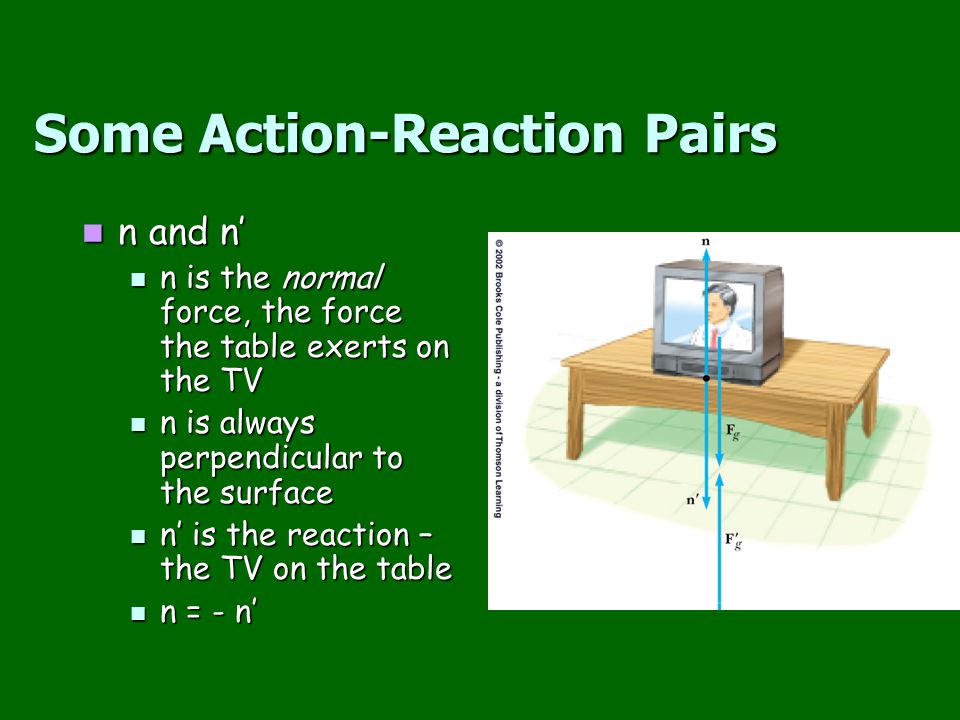{kind=link}
{kind=link}
{kind=link}
{kind=link}
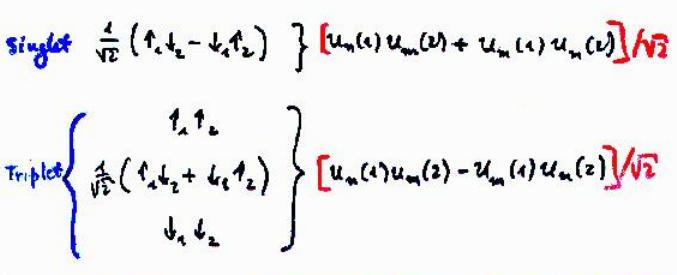{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
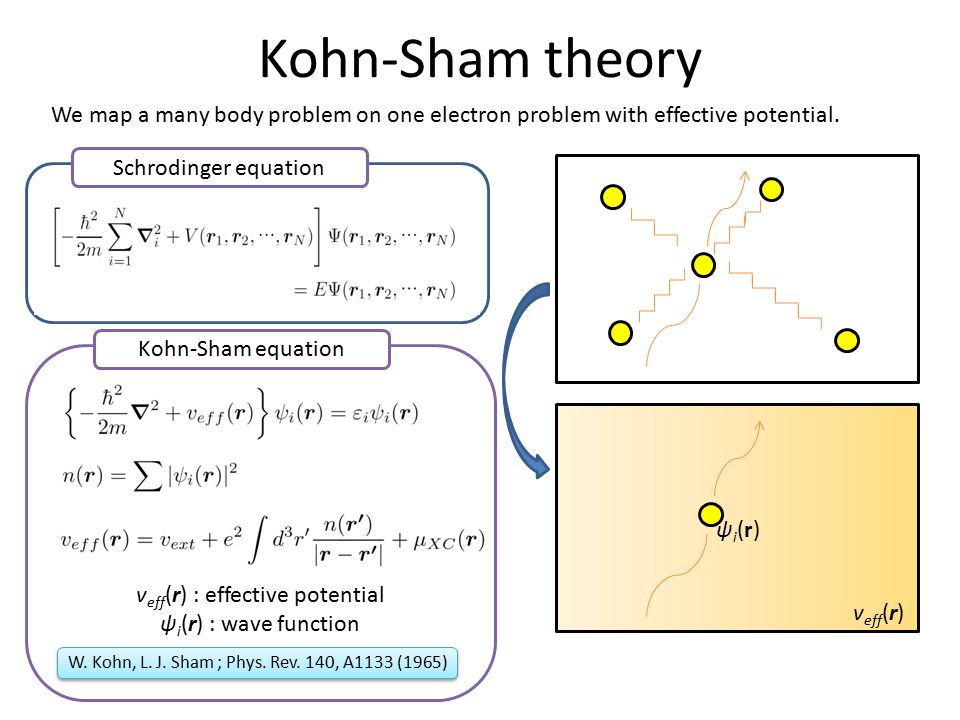{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
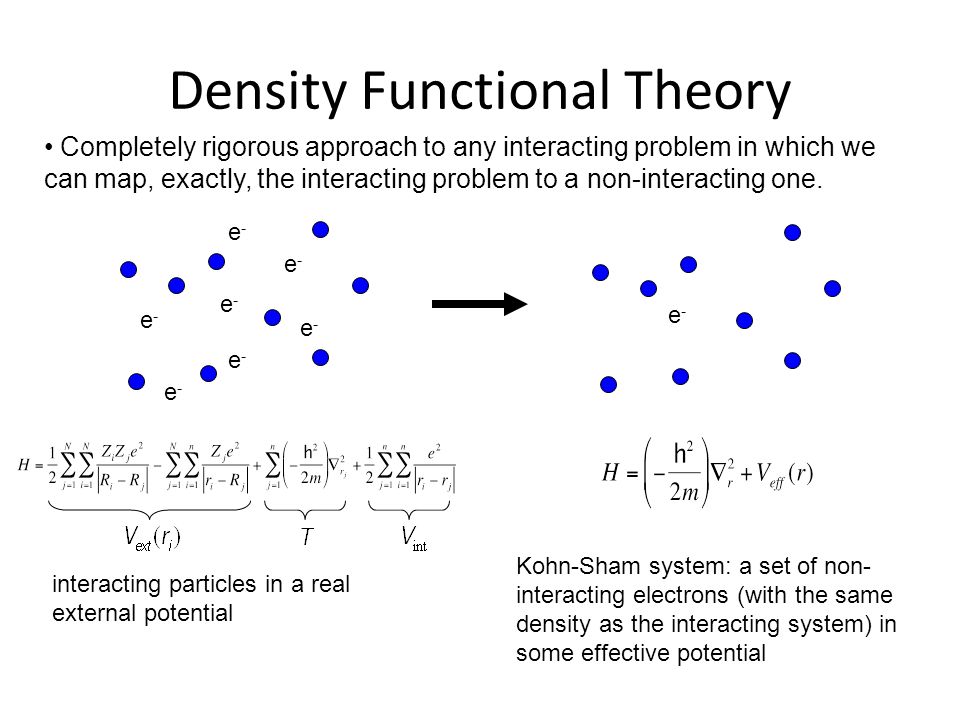{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
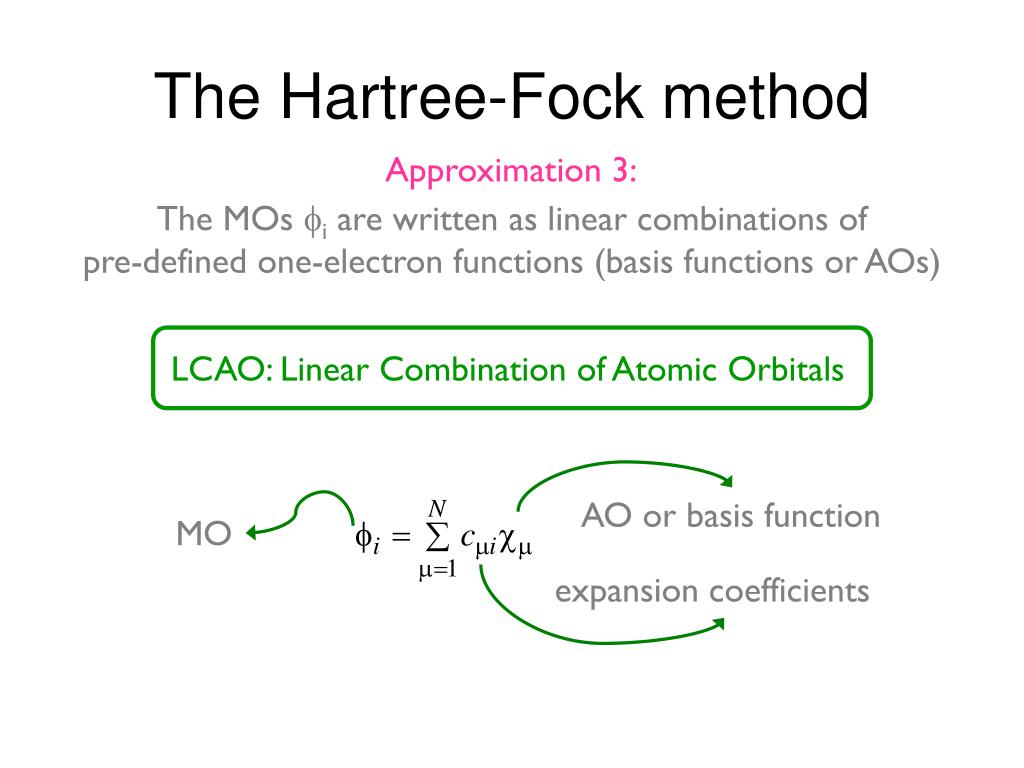{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
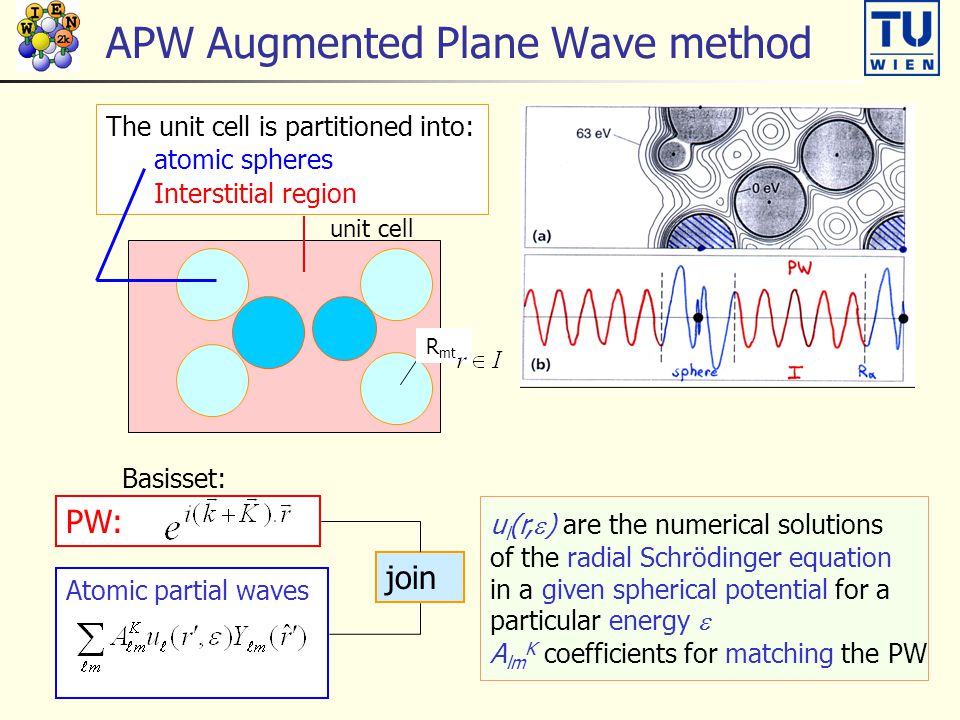{kind=link}
{kind=link}
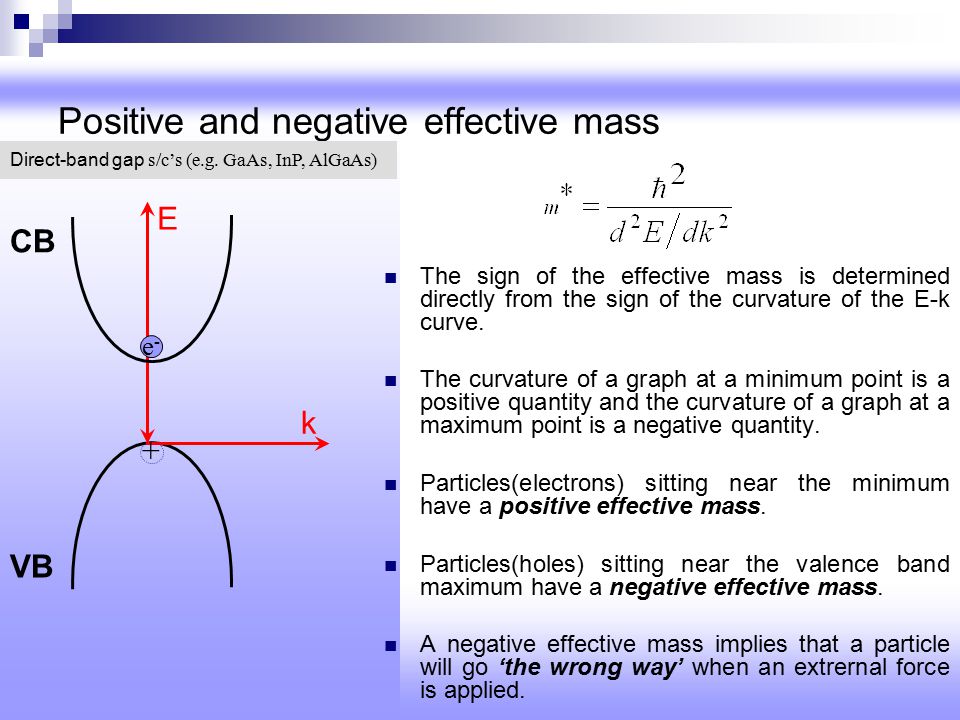{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}